ABSTRACT
- Transthyretin amyloid (ATTR) cardiomyopathy is a progressive disease caused by the infiltration of ATTR fibrils in the myocardium. Although it is a rare disease, ATTR cardiomyopathy is an important cause of heart failure with preserved ejection fraction, and its incidence is increasing due to improved diagnostic imaging tools. There has been a breakthrough in the field of transthyretin amyloidosis, which opens a new therapeutic door for the patients. In this review, an overview of tafamidis therapy in ATTR cardiomyopathy with recent results from clinical trials will be discussed.
-
Keywords: Amyloid; Cardiomyopathy; Restrictive; Tafamidis
INTRODUCTION
- Transthyretin (TTR) amyloid (ATTR) cardiomyopathy is a rare, progressive, fatal disease and is currently considered to be an underdiagnosed cause of heart failure (HF).1)2) ATTR cardiomyopathy is caused by the deposition of TTR protein in the myocardium. The exact prevalence of ATTR cardiomyopathy is uncertain; however, its incidence is increasing due to improvements in the diagnostic imaging tools and therapeutic measures. ATTR cardiomyopathy is classified into 2 subtypes: hereditary and wild-type ATTR cardiomyopathy. The prevalence of hereditary ATTR, which occurs through autosomal dominant inheritance, the prevalence varies according to the geographic regions. In endemic areas, some mutations are reported to range between 1 in 1,000,000 to 1 in 100,000.3)4) For wild-type ATTR, recent data suggest that in up to 10% of elderly patients with HF with preserved ejection fraction, the cause is wild-type ATTR cardiomyopathy.5)
- Tafamidis is a disease-modifying drug for the treatment of wild-type and hereditary ATTR cardiomyopathy, which terminates the amyloidogenesis of TTR protein by stabilizing TTR tetramers. This review will focus on the mechanisms of action and results of clinical trials with tafamidis.
DATA SELECTION
- PubMed (1966 to December 2020) and ClinicalTrials.gov were searched using the following terms: tafamidis, Vyndaqel, and Vyndamax. Articles regarding tafamidis and ATTR cardiomyopathy were reviewed.
AMYLOIDOGENESIS OF TTR PROTEIN
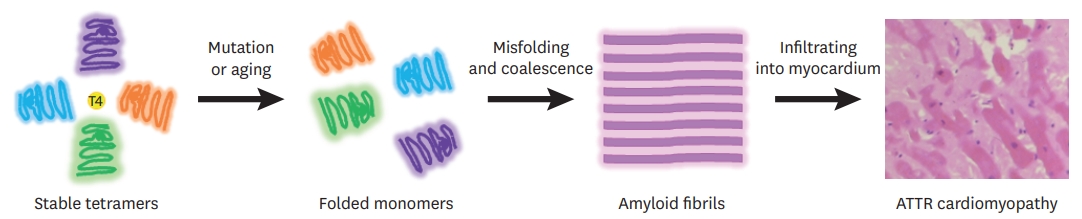
- TTR protein exists mainly as a tetramer, and each monomer is composed of 127 amino acids, forming extensive β-sheet structures.4)5) The TTR gene is located on chromosome 18, and the TTR protein is usually synthesized from the liver also produced by the choroid plexus and retinal pigmented epithelial cells. TTR proteins mainly transport the retinol-binding protein vitamin A complex and 15–20% of serum thyroxine.4)6) TTR protein is stable in its tetramer form, but when dissociated into unstable monomers, they undergo misfolding and aggregate into amyloid fibrils (Figure 1).7)8) Dissociation of tetramers into monomers is considered to be a rate-limiting process in TTR amyloidogenesis. A single mutation or aging process reduces the stability of TTR tetrameric forms and causes dissociations into the monomeric forms.9) Over 140 amyloidogenic mutations have been identified in hereditary ATTR, which are inherited as autosomal dominant traits with incomplete penetrance.10)11) In wild-type ATTR, the cause for dissociation of wild-type proteins into monomers and aggregation into amyloid fibril in the absence of mutations, is unknown; however, it is assumed to involve aging-associated oxidative stress.12)13) When TTR amyloid fibrils deposit into the organs, clinical manifestations are seen. In ATTR, the heart and nerves are mainly involved. In hereditary ATTR, the degrees of cardiac and nerve involvement vary according to the mutations. The common clinical presentation of ATTR cardiomyopathy is summarized in Table 1 and Figure 2.
CLINICAL PHARMACOLOGY
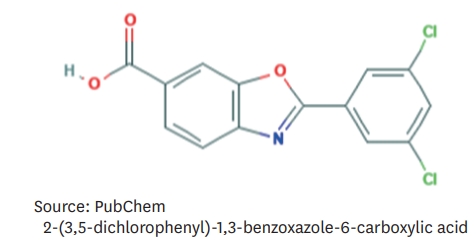
- With the finding that TTR tetramers are kinetically stabilized by the binding of thyroxine or retinol-binding protein, attempts were made to develop small molecules that bind to TTR tetramers to prevent their dissociation into TTR monomers. These molecules are referred to as TTR stabilizers. Tafamidis is a TTR stabilizer that binds to the thyroxine-binding sites of TTR tetramers.13) Tafamidis is a member of the class of 1,3-benzoxazoles (Figure 3)14) and lacks non-steroidal anti-inflammatory (NSAID) activity. This is important because previous TTR stabilizers with NSAID activity, such as diflunisal, were associated with gastrointestinal, renal, and cardiac adverse effects. Therefore, they were not well-tolerated in HF patients with ATTR cardiomyopathy.15)16) Tafamidis has a low toxicity profile and good oral bioavailability.15)
- According to a healthy volunteer pharmacokinetic analysis, there is no metabolic induction or inhibition after the administration of tafamidis. The median time to reach the maximum concentration is 2 hours, and the mean half-life is 59 hours.13-16) Pharmacokinetics showed that similar renal excretion clearance between the groups with creatinine clearance <80 and >80 mL/min. In patients with moderate hepatic impairment, a 40% decrease in systemic exposure was observed, possibly due to the increased amount of unbound tafamidis.17) Nevertheless, dosage adjustment is not necessary because TTR production from the liver is also decreased in patients with moderate hepatic dysfunction. Approximately 19% slower clearance was observed in subjects aged >65 years; however, dose adjustment is not required in elderly patients.15)
- Tafamidis stabilizes both wild-type TTR protein and mutant TTR protein regardless of the mutant type. In wild-type TTR treated with tafamidis, a 73% decrease in the tetramer dissociation rate was observed, and the rate of dissociation showed a strong correlation with the concentration of tafamidis in plasma.1) A previous study with different mutation TTR protein variants showed that although TTR protein variants exhibited different thermodynamic and kinetic stabilities, tafamidis consistently stabilized TTR tetramers regardless of the mutation variability.13)
CLINICAL OUTCOMES AND EFFICACY FROM PREVIOUS CLINICAL TRIALS
- The efficacy of tafamidis in ATTR cardiomyopathy was first described in 2019 as an openlabel, single treatment arm study.18) In this phase 2 study, 35 patients with hereditary (n=4, Val122Ile mutations) and wild-type (n=31) ATTR cardiomyopathy patients were treated with 20 mg tafamidis for 12 months. TTR stabilization was the primary endpoint, and clinical outcomes, including mortality and hospitalization, were evaluated. After 6 weeks of tafamidis treatment, TTR was effectively stabilized in 30 of 31 patients. During the 12 months of the study, 22.6% of the patients were hospitalized due to cardiovascular events, and 2 patients died. N-terminal prohormone brain natriuretic peptide (NT-proBNP) levels did not significantly increase during the study period. Troponin I and T were increased during the study period, and no clinically relevant echocardiographic changes were noted.19) Although tafamidis showed effective stabilization of TTR protein with generally good tolerability, the clinical benefits could not be adequately assessed due to the open-label design and short follow-up period.
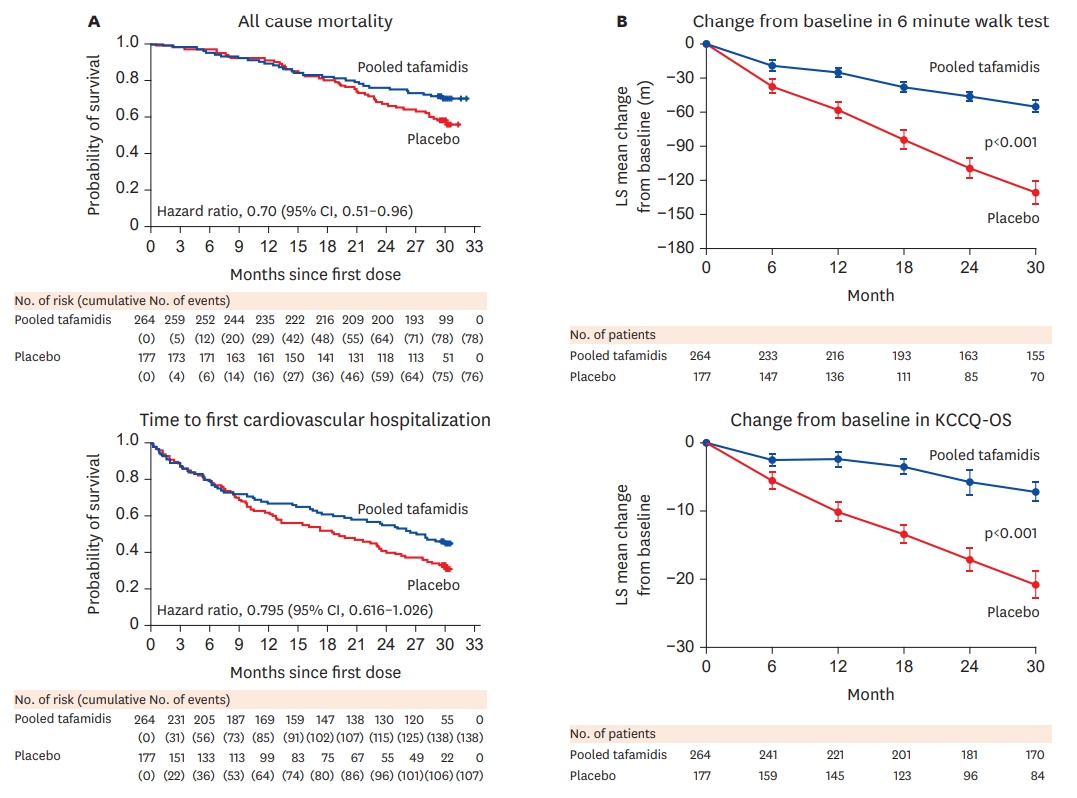
- In 2019, the results of the Transthyretin Amyloidosis Cardiomyopathy Clinical Trial reported clinical benefits of tafamidis treatment in ATTR cardiomyopathy. A total of 441 patients with ATTR cardiomyopathy were randomly assigned in a 2:1:2 ratio to receive 80 mg of tafamidis meglumine, 20 mg of tafamidis meglumine, or placebo for 30 months.20) The primary endpoints of this multi-center, international, double-blind, placebo-controlled, phase 3 trial included hierarchial analysis of all-cause mortality and the frequency of cardiovascular-related hospitalizations according to the Finkelstein–Schoenfeld method. In this trial, patients with New York Heart Association (NYHA) class IV HF, estimated glomerular filtration rate less than 24 mL/min/1.73 m2, liver enzymes exceeding twice the upper limit of the normal ranges, or patients with severe malnutrition (modified body mass index <600) were excluded. The median age of patients enrolled in the ATTR-ACT trial was 75 years, and the subjects were predominantly male (91% males in the tafamidis arm, 89% males in the placebo arm). Most patients had mild HF symptoms at baseline, with more than 50% of the enrolled patients classified as NYHA class II. Both arms included a higher proportion of wild-type ATTR cardiomyopathy than hereditary ATTR cardiomyopathy (76.1% and 75.7% in tafamidis and placebo groups, respectively). During the 30 months of followup, all-cause mortality was significantly lower in the tafamidis group than in the placebo group (Figure 4A) (29.5% vs. 42.9%; hazard ratio [HR], 0.70; 95% confidence interval [CI], 0.51–0.96). Cardiovascular-related hospitalizations were lower (0.48 vs. 0.70 per year; 95% CI, 0.56–0.81) among patients treated with tafamidis. The number of patients required to be treated to prevent 1 event of all-cause death for 30 months of treatment was 8. Tafamidis also reduced the decline in the distance covered during the 6-minute test (mean±standard error [SE], 75.68±9.24) and a decline in the functional capacity, as measured by the Kansas City Cardiomyopathy Questionnaire–Overall Summary score (mean±SE, 13.65±2.13) (Figure 4B). The decrease in the decline of distance covered during the 6-minute walk test and the reduced decline in functional capacity was seen as early as after 6 months of tafamidis treatment, while the reduction in all-cause mortality was observed after 18 months of treatment with tafamidis. In the subgroup analysis, the clinical benefit of tafamidis was consistent regardless of the TTR genotypes (hereditary vs. wild-type) or doses of tafamidis. Patients with baseline NYHA class IIII showed higher cardiovascular-related hospitalizations in the tafamidis group than in the placebo group. This finding is important. Since tafamidis is a kinetic stabilizer, it cannot reverse or reduce the already deposited TTR amyloid fibrils. Therefore, the clinical benefit of tafamidis is likely to be more profound in the earlier stages of ATTR cardiomyopathy. In this trial, the safety profiles of tafamidis meglumine 20 mg, 80 mg, and placebo were similar. Permanent discontinuation of tafamidis or placebo as a result of adverse events was more frequent in the placebo group.21) Previously reported adverse events in ATTR polyneuropathy patients taking tafamidis were diarrhea and urinary tract infection. In the ATTR-ACT trial, these adverse events were less common in the tafamidis group than in the placebo group. After 30 months of the study period, the ATTR-ACT trial was extended to investigate the optimal doses of tafamidis (80 mg vs. 20 mg).22) On completion of the 30-month double-blind study, patients were enrolled in a long-term extension (LTE) study and treated with tafamidis meglumine for an additional 60 months. Patients who had received placebo in the first phase of ATTR-ACT were re-randomized to receive either 80 or 20mg tafamidis meglumine in the LTE (in a 2:1 ratio; stratified by TTR genotype [hereditary vs. wild-type ATTR]). All-cause mortality with tafamidis meglumine 80mg compared with 20mg was assessed over a longer duration of treatment by combining data from the ATTRACT (median follow-up 30months) with LTE (median follow-up, 51months). Patients who were assigned to tafamidis meglumine 80 mg were significantly older, had more advanced NYHA class, higher NT-proBNP levels, and more functionally advanced HF than patients who were assigned to tafamidis 20 mg. All-cause mortality vs. placebo was reduced with tafamidis meglumine 80mg (HR, 0.690; 95% CI, 0.487–0.979; p=0.038) and 20mg (HR, 0.715; 95% CI, 0.450–1.137; p=0.156). There was a significantly greater survival benefit with tafamidis meglumine 80 than with tafamidis 20mg (HR, 0.700; 95% CI, 0.501–0.979; p=0.0374). After adjustment for age, NT-proBNP, and 6-minute walk test, the survival benefit was greater in patients treated with tafamidis meglumine 80 mg (43% after adjustment of all covariates vs. 30% without adjustment).22) In the LTE study, tafamidis meglumine 80 mg and 20 mg were generally well tolerated and had a comparable safety profile. No patients required reduction in doses due to treatment-emergent adverse events.
- Based on the ATTR-ACT and LTE studies, the recommended dosage for ATTR cardiomyopathy is tafamidis meglumine 80 mg (Vyndaqel®, 4 tablets of 20 mg capsule) orally once daily or tafamidis 61 mg (Vyndamax®, one 61mg capsule) orally once daily.23) Tafamidis 61 mg is a new, single capsule formulation bioequivalent to tafamidis meglumine 80 mg and was developed for patient convenience. In the LTE study, patients receiving tafamidis meglumine 80 mg were transited to tafamidis 61 mg after July 2018.
SAFETY PROFILE
- Tafamidis is generally well-tolerated. There was a concern that tafamidis might decrease the serum concentration of total thyroxine; however, there has no report of hypothyroidism in patients taking tafamidis.15) Most of the adverse events reported in the previous clinical trials in both ATTR cardiomyopathy and polyneuropathy are similar to the symptoms of the underlying disease. This is also reflected by the higher incidence of treatment-related adverse events in the placebo group in the ATTR-ACT trial.
CONCLUSION
- Tafamidis is currently the only approved drug by the United States Food and Drug Administration to treat ATTR cardiomyopathy. Tafamidis is generally well tolerated, and the clinical benefits are more significant in patients with mild HF symptoms, which highlights the importance of early diagnosis and treatment. Although tafamidis 61 mg (Vyndamax®) is not yet available in Korea, patients with ATTR cardiomyopathy can finally be treated with evidence-based medication that can change the prognosis of the disease. In addition, there are other disease-modifying drugs that are likely to benefit patients with ATTR cardiomyopathy. The emerging drugs suppress TTR protein synthesis in the liver through small interfering RNA (Patisiran)24) or antisense oligonucleotide (irinotecan).25) These drugs are in the final phases of clinical trials. Along with tafamidis, the upcoming promising agents will offer more therapeutic options for patients with ATTR cardiomyopathy.
- Since ATTR cardiomyopathy is a progressive disease, the current article explored the longterm cost-effectiveness of tafamidis for treatment.26) Based on prices in the United States, treating all eligible ATTR cardiomyopathy patients with tafamidis (estimated n=120,000) was estimated to increase the annual healthcare spending by $32.3 billion.26) On analyzing whether the efficacy of tafamidis could be limited by the high costs, it was concluded that a 92.6% price reduction would be necessary to make tafamidis cost-effective at $100,000/quality-adjusted life-year. Despite concerns, tafamidis is a breakthrough in treating patients with ATTR cardiomyopathy; it has good tolerability and proven clinical benefits.
ARTICLE INFORMATION
-
Conflict of Interest
The authors have no financial conflicts of interest.
-
Author Contributions
Conceptualization: Kim D ; Resources: Jeon ES; Supervision: Choi JO, Jeon ES; Visualization: Kim D ; Writing - original draft: Kim D; Writing - review & editing: Choi JO.
Figure 1.
Schematic diagram of TTR amyloidogenesis. Each monomer consists of 127 amino acids with 1 alpha helix and 8 beta strands. Tetrameric TTR protein has 2 binding sites in a central channel called the T4 pocket; however, the T4 hormone preferentially binds to only 1 of the 2 sites. Stability of TTR tetramers is reduced by mutation or aging processes. As a result, TTR tetramers dissociate into dimeric or monomeric forms. Unstabilized monomers undergo misfolding and aggregation, forming amyloid fibrils.
ATTR = transthyretin amyloidosis; TTR = transthyretin.
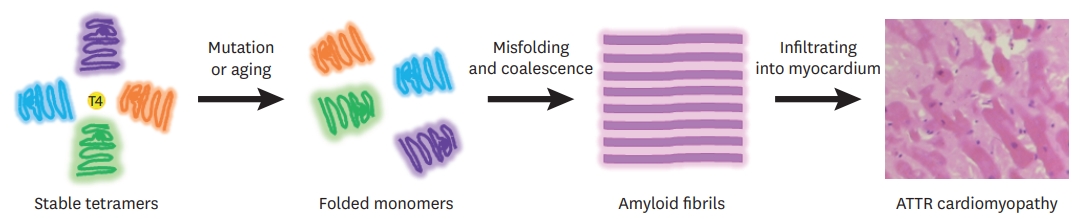
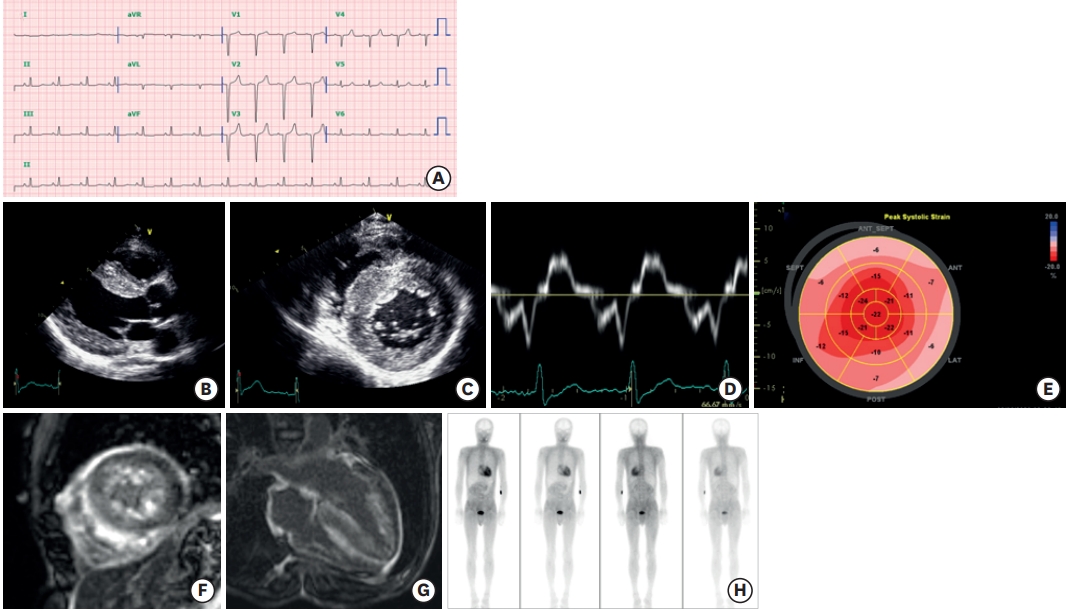
Figure 2.
Clinical features of ATTR cardiomyopathy. A 47-year-old man visited the cardiology clinic complaining of dyspnea on exertion and bilateral pitting edema. The jugular vein was prominent. He had undergone surgery for bilateral carpal tunnel syndrome 3 years ago. He also complained of frequent episodes of diarrhea. The electrocardiography showed a pseudo-infarct pattern (A) and N-terminal prohormone brain natriuretic peptide was 3,600 pg/mL. Transthoracic echocardiography showed a thickened myocardium (15 mm at the septum), a relatively small left ventricle (end-diastolic dimension 45 mm), decreased e′ velocity (4.2 cm/s) with apical sparing of longitudinal strain (B-E). Cardiac magnetic resonance imaging revealed multifocal late gadolinium enhancement with subendocardial ring enhancement (F, G) Tc-99m-3,3-diphosphono-1,2-propanodicarboxylic acid scan showed grade 3 cardiac uptake with increased radioactive uptake in the gastrointestinal tract as well (H). Gene analysis was performed, and Asp58Val mutation was discovered. He was diagnosed with ATTR with involvement of the heart, gastrointestinal tract, and peripheral nerves.
ATTR = transthyretin amyloidosis.
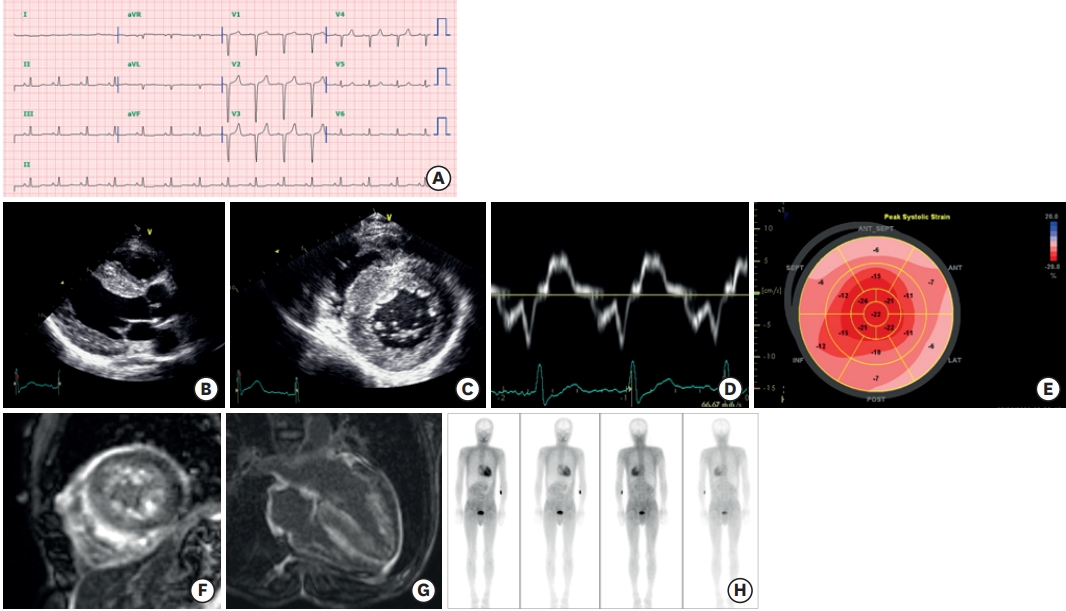
Figure 3.Molecular structure of tafamidis.14)
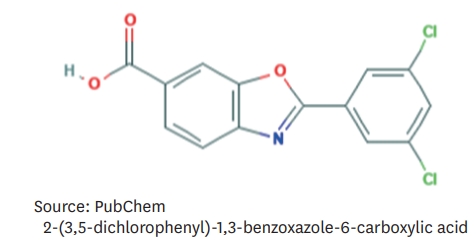
Figure 4.
Clinical outcome in ATTR-ACT trial (modifications of Figures 2 and 4 from 20)) Reduction of all-cause mortality as well as reduction of functional decline was noted in patients treated with tafamidis when compared to those who received placebo.
KCCQ-OS = Kansas City Cardiomyopathy Questionnaire–Overall Summary.

Table 1.Clinical presentation of ATTR cardiomyopathy
|
Clinical presentations |
|
Echocardiography |
Unexplained increased in wall thickness (>12 mm) with non-dilated LV |
|
Thickening of RV free walls, valves, or interatrial septum |
|
Small pericardial effusion |
|
Reduced LV GLS with apical sparing pattern despite preserved ejection fraction |
|
ECG |
Pseudo infarct-pattern or low voltage*
|
|
Labs |
Mild increase in troponin levels on repeated occasions |
|
Cardiac presentation |
Atrioventricular block in presence of increased LV wall thickness |
|
Unexplained conduction block needing pacemaker |
|
Elderly HF with preserved ejection fraction refractory to conventional HF therapy |
|
Intolerance to beta blocker, ACEi or ARB |
|
Self-improving hypertension |
|
Low normal blood pressure with previous history of hypertension |
|
Restrictive hemodynamic profile |
|
Extracardiac manifestation |
Autonomic signs and symptoms (orthostatic hypotension, alternating constipation/diarrhea, sweating abnormalities) associated with peripheral neuropathy |
|
Musculoskeletal symptoms: carpal tunnel syndrome, particularly if bilateral, spinal stenosis |
REFERENCES
- 1. Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S, Lewis WD, Obici L, Planté-Bordeneuve V, Rapezzi C, Said G, Salvi F. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 2013;8:31. ArticlePubMedPMC
- 2. Kato-Motozaki Y, Ono K, Shima K, Morinaga A, Machiya T, Nozaki I, Shibata-Hamaguchi A, Furukawa Y, Yanase D, Ishida C, Sakajiri K, Yamada M. Epidemiology of familial amyloid polyneuropathy in Japan: identification of a novel endemic focus. J Neurol Sci 2008;270:133–40.ArticlePubMed
- 3. Rapezzi C, Quarta CC, Obici L, Perfetto F, Longhi S, Salvi F, Biagini E, Lorenzini M, Grigioni F, Leone O, Cappelli F, Palladini G, Rimessi P, Ferlini A, Arpesella G, Pinna AD, Merlini G, Perlini S. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J 2013;34:520–8.ArticlePubMed
- 4. Hagen GA, Elliott WJ. Transport of thyroid hormones in serum and cerebrospinal fluid. J Clin Endocrinol Metab 1973;37:415–22.PubMed
- 5. Rappley I, Monteiro C, Novais M, Baranczak A, Solis G, Wiseman RL, Helmke S, Maurer MS, Coelho T, Powers ET, Kelly JW. Quantification of transthyretin kinetic stability in human plasma using subunit exchange. Biochemistry 2014;53:1993–2006.ArticlePubMed
- 6. Liz MA, Mar FM, Franquinho F, Sousa MM. Aboard transthyretin: from transport to cleavage. IUBMB Life 2010;62:429–35.ArticlePubMed
- 7. Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol 2019;15:387–404.ArticlePubMed
- 8. Kelly JW. Amyloid fibril formation and protein misassembly: a structural quest for insights into amyloid and prion diseases. Structure 1997;5:595–600.ArticlePubMed
- 9. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003;349:583–96.ArticlePubMed
- 10. Rowczenio DM, Noor I, Gillmore JD, Lachmann HJ, Whelan C, Hawkins PN, Obici L, Westermark P, Grateau G, Wechalekar AD. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat 2014;35:E2403–12.ArticlePubMed
- 11. Purkey HE, Dorrell MI, Kelly JW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci U S A 2001;98:5566–71.ArticlePubMedPMC
- 12. Zhao L, Buxbaum JN, Reixach N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry 2013;52:1913–26.ArticlePubMed
- 13. Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW; Labaudinière R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A 2012;109:9629–34.ArticlePubMedPMC
- 14. National Center for Biotechnology Information. PubChem compound summary for CID 11001318, tafamidis [Internet] Bethesda, MD: National Center for Biotechnology Information; 2020 [cited 2020 Dec 31]. Available from https://pubchem.ncbi.nlm.nih.gov/compound/Tafamidis
- 15. Coelho T, Merlini G, Bulawa CE, Fleming JA, Judge DP, Kelly JW, Maurer MS, Planté-Bordeneuve V, Labaudinière R, Mundayat R, Riley S, Lombardo I, Huertas P. Mechanism of action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol Ther 2016;5:1–25.ArticlePubMedPMC
- 16. Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006;13:236–49.ArticlePubMed
- 17. Schmidt HHJ. Tafamidis for the treatment of transthyretin-associated familial amyloid polyneuropathy. Expert Opin Orphan Drugs 2013;1:837–45.Article
- 18. Maurer MS, Sultan MB, Rapezzi C. Tafamidis for transthyretin amyloid cardiomyopathy. N Engl J Med 2019;380:196–7.Article
- 19. Maurer MS, Grogan DR, Judge DP, Mundayat R, Packman J, Lombardo I, Quyyumi AA, Aarts J, Falk RH. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail 2015;8:519–26.PubMed
- 20. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB; Rapezzi CATTR-ACT Study Investigators. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007–16.ArticlePubMed
- 21. Manion C, Sharma UC. Tafamidis for transthyretin amyloid cardiomyopathy. N Engl J Med 2019;380:196–7.Article
- 22. Damy T, Garcia-Pavia P, Hanna M, Judge DP, Merlini G, Gundapaneni B, Patterson TA, Riley S, Schwartz JH, Sultan MB, Witteles R. Efficacy and safety of tafamidis doses in the tafamidis in transthyretin cardiomyopathy clinical trial (ATTR-ACT) and long-term extension study. Eur J Heart Fail 2020;Oct 18 [Epub ahead of print]. https://doi.org/10.1002/ejhf.2027.Article
- 23. Park J, Egolum U, Parker S, Andrews E, Ombengi D, Ling H. Tafamidis: a first-in-class transthyretin stabilizer for transthyretin amyloid cardiomyopathy. Ann Pharmacother 2020;54:470–7.ArticlePubMed
- 24. Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, Perez J, Chiesa J, Warrington S, Tranter E, Munisamy M, Falzone R, Harrop J, Cehelsky J, Bettencourt BR, Geissler M, Butler JS, Sehgal A, Meyers RE, Chen Q, Borland T, Hutabarat RM, Clausen VA, Alvarez R, Fitzgerald K, Gamba-Vitalo C, Nochur SV, Vaishnaw AK, Sah DW, Gollob JA, Suhr OB. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 2013;369:819–29.ArticlePubMed
- 25. Benson MD, Ackermann EJ, Monia BP. Treatment of transthyretin cardiomyopathy with a TTR-specific antisense oligonucleotide (IONIS-TTRRx). Amyloid 2017;24:134–5.ArticlePubMed
- 26. Kazi DS, Bellows BK, Baron SJ, Shen C, Cohen DJ, Spertus JA, Yeh RW, Arnold SV, Sperry BW, Maurer MS, Shah SJ. Cost-effectiveness of tafamidis therapy for transthyretin amyloid cardiomyopathy. Circulation 2020;141:1214–24.ArticlePubMedPMC
Citations
Citations to this article as recorded by

- A Comprehensive Review on Chemistry and Biology of Tafamidis in
Transthyretin Amyloidosis
Monali B. Patil, Piyush Ghode, Prashant Joshi
Mini-Reviews in Medicinal Chemistry.2024; 24(6): 571. CrossRef - Multimodal Imaging and Biomarkers in Cardiac Amyloidosis
Mi-Hyang Jung, Suyon Chang, Eun Ji Han, Jong-Chan Youn
Diagnostics.2022; 12(3): 627. CrossRef
 , Jin-Oh Choi, MD, PhD
, Jin-Oh Choi, MD, PhD

 PubReader
PubReader ePub Link
ePub Link Cite
Cite