ABSTRACT
- Nonalcoholic fatty liver disease (NAFLD), which has recently undergone a change in its definition and acronym to “metabolic dysfunction-associated fatty liver disease (MAFLD),” is clinically significant as an increasingly prevalent independent risk factor for cardiovascular diseases. Insulin resistance is considered to be a key mechanism in the development and progression of NAFLD/MAFLD, and fatty liver disease itself may exacerbate insulin resistance. In this review, we describe the mechanisms underlying the interaction between insulin resistance and fatty liver, and we summarize the therapeutic attempts based on those mechanisms.
-
Keywords: Insulin resistance; Nonalcoholic fatty liver disease; Metabolic dysfunction-associated fatty liver disease; Inflammation; Metabolic syndrome
INTRODUCTION
- The diagnosis of nonalcoholic fatty liver disease (NAFLD) requires the presence of imaging or histologically documented steatosis (>5%), and as a “condition of exclusion,” it is necessary to rule out secondary causes that induce such steatosis, such as alcohol intake, viral infection, drugs, and genetic factors [1,2]. However, based on the argument that the term “NAFLD” does not reflect the diversity and heterogeneity of the disease, a new definition was proposed along with a new term, “metabolic dysfunction-associated fatty liver disease (MAFLD)” [3]. MAFLD is a “diagnosis of inclusion” that refers to a condition in which various liver diseases, including those induced by alcohol, may coexist. In particular, diabetes and insulin resistance (IR) are becoming important diagnostic criteria [4]. However, these two terms are often used interchangeably without an exact distinction, and the results of previous studies and our extant understanding of fatty liver are all based on the preexisting definition of NAFLD. In this review, the term “NAFLD” will continue to be used when describing previous research results.
- The global prevalence of NAFLD is approximately 25%, despite regional variation, and its prevalence is gradually increasing, which is related to the increasing prevalence of obesity [5–7]. NAFLD patients have a higher mortality rate than those without NAFLD, and NAFLD is clinically significant as an independent risk factor for cardiovascular disease (CVD) [8–11]. This review summarizes the molecular pathophysiology of IR, a key mechanism of NAFLD, which is emerging as an important culprit of CVD [12,13], and discusses therapeutic attempts based on this theoretical background.
OBESITY, INFLAMMATION, AND IR
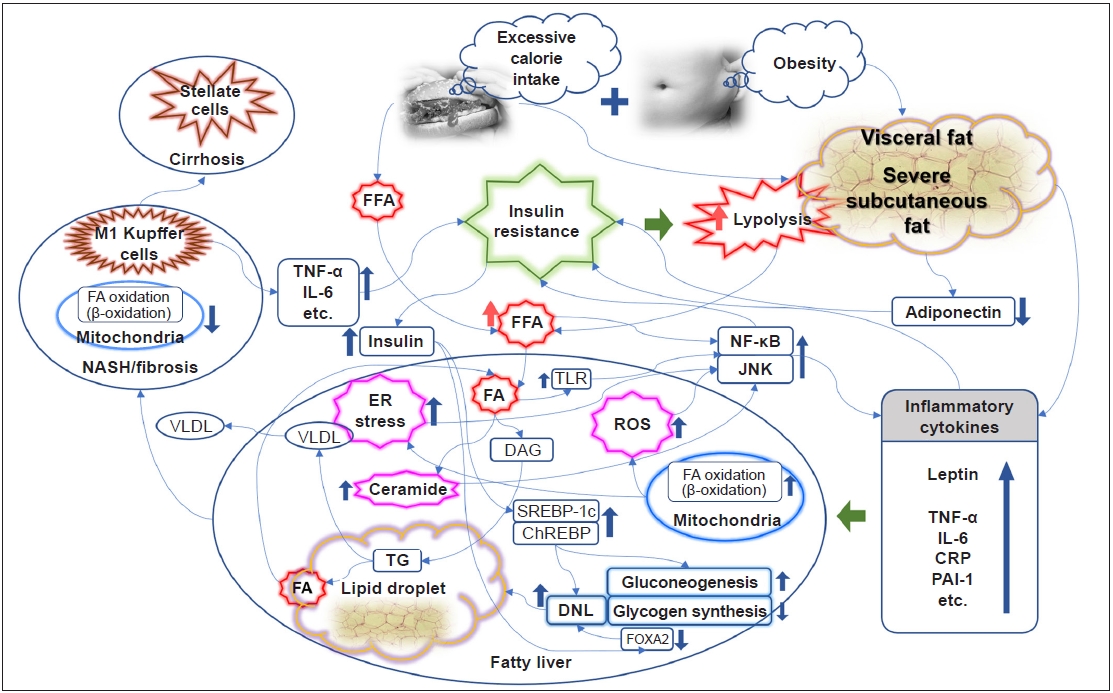
- It is well known that obesity promotes systemic inflammatory conditions and is thus closely related to the induction of IR [14]. In particular, abdominal visceral fat is associated with peripheral and hepatic IR in patients with type 2 diabetes, and excessive subcutaneous fat in men is also associated with hepatic and peripheral IR [15]. A comparative study found that abdominal fat accumulation was correlated with IR, but subcutaneous fat accumulation was correlated with the level of leptin [16]. Thus, IR may be affected by fat distribution, especially abdominal obesity, rather than simple obesity. The levels of adiponectin, an adipokine secreted from adipocytes, are inversely associated with the amount of fat in the abdomen and liver, which is closely related to hepatic and peripheral IR [17,18]. Obesity and its subsequent activation of proinflammatory pathways, including tumor necrosis factor-α (TNF-α), C-reactive protein, interleukin-6 (IL-6), plasminogen activator inhibitor-1, and leptin, are considered an important causative element in the pathophysiology of IR [14,19]. A rat-based study identified for the first time that adipocytes express the inflammatory cytokine TNF-α, and in particular, TNF-α levels are further increased in the obese state [20]. Consistent results have also been reported in humans [21]. Obesity is also associated with elevated IL-6 levels [22]. The elevation of TNF-α and IL-6 is thought to have a causal relationship with IR and type 2 diabetes [23]. Endoplasmic reticulum stress, reactive oxygen species, and ceramide induced in conditions of obesity or excessive nutrient intake lead to activation of the nuclear factor-κB (NF-κB) pathway and the c-Jun NH2-terminal kinase (JNK) pathway, resulting in an increase in inflammatory cytokines that inhibit insulin signaling [19,24–27]. In a knockout mouse model, inhibition of JNK1 and the inhibitor of NF-κB kinase β (IKK-β), which activates NF-κB, ameliorated IR locally (liver) or systemically [28,29]. The anti-inflammatory mechanism of insulin is well known [30,31]. As a mechanism of mutual influence, IR caused by activation of the inflammatory pathway further exacerbates inflammation, resulting in a vicious cycle (Fig. 1) [32].
IR AND HEPATIC STEATOSIS/STEATOHAPATITIS
- NAFLD is a disease with a diverse spectrum that leads to nonalcoholic steatohepatitis (NASH) and cirrhosis, and is closely related to the development of type 2 diabetes, as well as CVD [33]. The association between NAFLD and hepatic IR, as well as systemic or adipocyte IR, has been well documented in previous studies [34–36], and the relationship may be independent of the degree of visceral fat [37,38]. In research from recent decades, IR has been considered a key mechanism responsible for the development and progression of NAFLD [33,39,40]. The hepatic fatty acid supply, the source of triacylglycerol (TG) synthesis, is largely divided into diet, lipolysis, and hepatic de novo lipogenesis (DNL) [36]. In addition to the dietary fatty acid supply from a high-fat diet, a high-carbohydrate diet also promotes DNL in the liver [41]. The most important concern is that the largest source of TG synthesis in patients with NAFLD under dietary control is free fatty acids (FFAs) from lipolysis in adipocytes [42]. Insulin has a strong anabolic action, especially insofar as it promotes the synthesis of adipocytes while inhibiting lipolysis. The suppression of the antilipolysis effect of insulin under IR conditions leads to the excessive production of FFAs, which enter the liver and accumulate [43,44]. Moreover, hyperinsulinemia under IR conditions also promotes DNL. In mouse models, hepatic adipogenesis was regulated according to the activity of sterol receptor binding protein 1c (SREBP-1c) [45,46], and increased insulin levels activate SREBP-1c [47]. As a result, elevated TG levels are used as another source of circulating FFAs, and during this process, very-low-density lipoprotein synthesis is also elevated. However, activation of the forkhead box protein A2 (FOXA2) promotes lipid metabolism and ketogenesis (i.e., fatty acid oxidation). FOXA2 is activated in low-insulin conditions, such as a fasting state, while remaining inactive in hyperinsulinemia with IR, promoting hepatic lipid accumulation (Fig. 1) [48].
- Through these various effects, IR induces early hepatic steatosis, which can be said to be an “adaptation process” for excessive FFA influx. As part of this adaptation process, hepatic mitochondrial respiratory rates are increased in obese individuals with IR only but no NASH [49]. Reducing the conversion to TG by inhibiting diacylglycerol O‑acyltransferase 2, a key enzyme in the conversion process from FFA to TG, causes excessive fatty acid oxidation through cytochrome P450 2E1 (CYP2E1) and increases oxidative stress. This leads to hepatocellular damage that worsens into steatohepatitis [50].
- Although IR is described as a major physiological cause of fatty liver, some hypotheses and evidence also suggest that fatty liver itself is one of the major causes of IR exacerbation under various conditions, such as a high-fat diet [36,51,52]. FFA influx into the liver induces abnormal increases in long-chain fatty acyl-CoA and diacylglycerol/TG, and increased levels of these substances induce the translocation of protein kinase C-δ (PKC-δ) from the cytosol to the plasma membrane, resulting in its activation. It is thought that this activated PKC-δ phosphorylates insulin receptor substrates and molecules in the insulin pathway, causing hepatic IR and increased glucose production in the liver [53]. FFAs also activate the IKK-β and JNK pathways, inducing IR via PKC-θ activation [54]. These NF-κB and JNK pathways are also activated by Toll-like receptor, which can be activated by fatty acid influx [55,56]. When SREBP-1c, which is activated in hyperinsulinemia and plays a role in promoting DNL, was overexpressed in mice, the homeostasis model assessment of IR (HOMA-IR), a metric for evaluating IR, increased [57]. Mice with fatty liver induced through the inhibition of fatty acid oxidation showed systemic IR [58]. A particularly interesting fact is that “hepatic mitochondrial flexibility (increased mitochondrial respiratory rates)” is lost in NASH, while hepatic IR and systemic inflammation further progress [49], leading to a vicious cycle (Fig. 1).
- In NAFLD with hepatic IR, the concept of pathway-specific hepatic insulin resistance was introduced to explain the paradoxical condition in which DNL is increased, whereas the inhibition of hepatic gluconeogenesis is rather impaired, even in hyperinsulinemia. That is, the insulin activation pathway protein kinase B/forkhead box protein O1 pathway is inhibited, whereas the SREBP-1c pathway is maintained and activated [59]. However, recently, the role of lipogenic substrates has been recognized as important in this process, and a recent review described research results showing that the activation of carbohydrate response element-binding protein (ChREBP) induces an increase in precursors of DNL and an increase in enzymes that aggravate hepatic steatosis, especially under exposure to lipogenic substrates (Fig. 1) [60].
- If conditions of IR and increased FFA influx persist, hepatic damage caused by reactive oxygen species induced by fatty acid oxidation and direct lipotoxicity accumulates. At this stage, hepatic macrophages (i.e., Kupffer cells), especially proinflammatory M1 Kupffer cells stimulated by Toll-like receptor ligands and interferon-γ, play an important role in the progression of NAFLD to fibrosis or NASH. These cells secrete inflammatory cytokines (TNF-α, IL-6, etc.) that induce hepatic and systemic IR (Fig. 1) [61,62].
- In actual clinical studies, the degree of IR evaluated by HOMA-IR could predict the current fibrosis stage and future fibrosis progression in patients with NAFLD [63–66]. Changes in HOMA-IR were even associated with changes in fibrosis status when evaluated by noninvasive fibrosis indices [67].
IR AND THERAPEUTIC POTENTIAL
- Based on the mechanisms of IR interconnected with the development and progression of NAFLD/MAFLD described so far, it can be inferred that a therapeutic approach focused on improving IR may be the key to treating NAFLD/MAFLD [1,2]. Clinically, improvements in hyperinsulinemia alone could actually reduce the risk of NAFLD in subjects without diabetes [39]. To date, regardless of obesity or diabetes, the only validated and approved treatment for NAFLD/MAFLD is lifestyle modification [1,2,68]: weight loss [69–73], calorie restriction [74,75], sustained exercise above a certain intensity [76–78], or a combination of these interventions [70,79]. Hepatic steatosis improved only with a weight loss of 5% or more, and a weight loss of 7% or more provided histological improvement such as inflammation and fibrosis. When diet was restricted to 30% or less than 1,000 kcal per day, hepatic steatosis and IR improvement could be obtained. In addition, exercising for 150 minutes or more per week or engaging moderate-intensity exercise (≥10 minutes) five or more times per week was found to lead to improvements in NAFLD and aminotransferase enzyme levels, regardless of body mass index or weight loss. In particular, incorporating exercise rather than diet restriction alone led to more weight loss and histological improvement.
- Pharmacological treatment may also be helpful, although it is limited to cases that have progressed to NASH or fibrosis and cases accompanied by complications such as diabetes. Among insulin sensitizers, metformin, a representative member of the biguanide class, improved aminotransferase levels and liver volume in NASH patients [80], and, in an open-label, randomized control trial, metformin showed better improvement in liver enzyme levels than vitamin E or dietary regimens. However, histological improvement could not be confirmed statistically [81]. A meta-analysis interpreting various other randomized controlled trials confirmed that metformin can help improve liver enzyme levels and IR, but does not guarantee histological improvement [82].
- Pioglitazone, which is the only approved and available thiazolidinedione in the United States and Europe excluding Korea, is an agonist of peroxisome proliferator-activated receptor-γ that has demonstrated histological improvement in patients with NASH with or without type 2 diabetes in many studies [83–86]. In those studies, administration of pioglitazone led to improvements in the NAFLD activity score and liver enzyme levels, as well as some improvement in fibrosis, compared to the control group. In a multicenter, prospective, open-label, exploratory clinical trial conducted in Korea, lobeglitazone also showed improvements in liver enzyme levels and hepatic steatosis evaluated by transient liver elastography [87]. Thiazolidinedione improves IR in various parts of the body, such as the skeletal muscle and fat, as well as the liver, and redistributes liver and visceral fat to the periphery. It also promotes fatty acid oxidation. The most common adverse event is an increase in body weight. Although some studies have reported that pioglitazone is associated with an elevated risk for bladder cancer [88], other studies have found no such associations [89].
- Vitamin E is expected to improve IR through its antioxidant effects, and it led to histological improvements in nondiabetic NASH patients in the Placebo for the Treatment of Nondiabetic Patients with Nonalcoholic Steatohepatitis (PIVENS) trial [84]. In this trial, 800 IU/day of vitamin E supplementation showed improvement in NAFLD activity score, hepatocellular ballooning, and lobular inflammation. In the Treatment of NAFLD in Children (TONIC) trial for children aged 8 to 17 years, vitamin E prescription did not show a statistically significant reduction in alanine aminotransferase and hepatic steatosis, but it was confirmed that hepatocellular ballooning improved, and the NASH disappearance rate was high [90]. A meta-analysis study based on randomized clinical trials has demonstrated the usefulness of vitamin E for NAFLD/NASH [91]. For NASH patients who are not obese, some societies may consider prescribing vitamin E under the premise that there is no diabetes and cirrhosis [68]. However, further research is still needed on the issue of long-term safety, including mortality or malignancy, and the consistency of effectiveness of this agent [92,93].
- Some published studies have demonstrated the effects of glucagon-like peptide-1 (GLP-1) agonists on NAFLD/NASH [94,95]. In NASH patients, when 1.8 mg of liraglutide was administered subcutaneously daily, the NASH remission rate was four times higher than in the placebo group (relative risk, 4.3) and the risk of fibrosis progression was five times lower (relative risk, 0.2) [94]. Moreover, when 0.1 to 0.4 mg of semaglutide was subcutaneously administered daily, the NASH remission rate was at least two times higher than in the placebo group, and in particular, the 0.4-mg administration group had a NASH remission rate that was more than three times higher (17% vs. 59%) [95]. Recent studies have suggested various metabolic benefits of sodium-glucose cotransporter 2 (SGLT2) inhibitors. In patients with type 2 diabetes who received empagliflozin (25 mg) daily for 24 weeks, compared to the placebo group, the decrease in intrahepatic fat mass as assessed by magnetic resonance imaging was 2.3 times greater, and the adiponectin level increased (36%). However, there was no change in insulin sensitivity [96]. In type 2 diabetes patients with NAFLD who received dapagliflozin (10 mg) daily for 12 weeks, the amount of intrahepatic fat was also significantly reduced compared to the placebo group (5.8±5.1 Hounsfield units vs. 0.5±6.1 Hounsfield units, P=0.006), but there were no significant changes in the levels of adipokines, such as adiponectin [97]. However, GLP-1 agonists and SGLT2 inhibitors are not yet officially recommended for the treatment of NAFLD/MAFLD [2,68].
- In addition to the agents mentioned above, new concepts of drugs are being proposed. In the case of elafibranor, a proliferator-activated receptor-α/δ agonist, histological improvement could not be confirmed in NASH patients, but improvements in HOMA-IR, FFA, and TG were identified in more severe inflammatory conditions [98]. Obeticholic acid, a farnesoid X receptor agonist, which can be expected to improve steatosis by inhibiting the expression of SREBP-1c and ChREBP, is currently undergoing a phase 3 clinical study. According to the data published so far, histological improvement was shown in NAFLD/NASH patients, but its safety was not yet proven [99,100].
CONCLUSIONS
- The clinical significance of NAFLD/MAFLD is increasingly being emphasized, as its prevalence increases, and there is currently a shift toward new definitions and concepts of this condition. IR is a key pathophysiological mechanism that can be both a cause and a consequence of NAFLD/MAFLD. Fatty liver is a representative phenotype of various metabolic diseases, and active early management and attention are needed given its role in increasing the risk of CVD.
ARTICLE INFORMATION
-
Ethical statements
Not applicable.
-
Conflicts of interest
The authors have no conflicts of interest to declare.
-
Funding
None.
-
Author contributions
Conceptualization: all authors; Supervision: WYL; Validation: WYL; Visualization: DJK; Writing–original draft: DJK; Writing–review & editing: all authors.
All authors read and approved the final manuscript.
Fig. 1.Link between insulin resistance and the development and progression of nonalcoholic fatty liver disease. ChREBP, carbohydrate response element-binding protein; CRP, C-reactive protein; DAG, diacylglycerol; DNL, de novo lipogenesis; ER, endoplasmic reticulum; FA, fatty acid; FFA, free fatty acid; FOXA2, forkhead box protein A2; IL-6, interleukin-6; JNK, c-Jun NH2-terminal kinase; NASH, nonalcoholic steatohepatitis; NF-κB, nuclear factor-κB; PAI-1, plasminogen activator inhibitor-1; ROS, reactive oxygen species; SREBP-1c, sterol receptor binding protein-1c; TG, triacylglycerol; TLR, Toll-like receptor; TNF-α, tumor necrosis factor-α; VLDL, very-low-density lipoprotein.
REFERENCES
- 1. European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. Obes Facts 2016;9:65–90.ArticlePubMedPMCPDF
- 2. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328–57.ArticlePubMedPDF
- 3. Eslam M, Sanyal AJ, George J; International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020;158:1999–2014.ArticlePubMed
- 4. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol 2020;73:202–9.ArticlePubMed
- 5. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 2011;34:274–85.ArticlePubMed
- 6. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease: meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73–84.ArticlePubMed
- 7. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11–20.ArticlePubMedPDF
- 8. Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006;44:865–73.ArticlePubMed
- 9. Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013;5:1544–60.ArticlePubMedPMC
- 10. Younossi Z, Henry L. Contribution of alcoholic and nonalcoholic fatty liver disease to the burden of liver-related morbidity and mortality. Gastroenterology 2016;150:1778–85.ArticlePubMed
- 11. Adams LA, Anstee QM, Tilg H, Targher G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 2017;66:1138–53.ArticlePubMed
- 12. Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med 2010;363:1341–50.ArticlePubMed
- 13. Bhatia LS, Curzen NP, Calder PC, Byrne CD. Non-alcoholic fatty liver disease: a new and important cardiovascular risk factor? Eur Heart J 2012;33:1190–200.ArticlePubMed
- 14. Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007;132:2169–80.ArticlePubMed
- 15. Miyazaki Y, Glass L, Triplitt C, Wajcberg E, Mandarino LJ, DeFronzo RA. Abdominal fat distribution and peripheral and hepatic insulin resistance in type 2 diabetes mellitus. Am J Physiol Endocrinol Metab 2002;283:E1135–43.ArticlePubMed
- 16. Cnop M, Landchild MJ, Vidal J, Havel PJ, Knowles NG, Carr DR, et al. The concurrent accumulation of intra-abdominal and subcutaneous fat explains the association between insulin resistance and plasma leptin concentrations: distinct metabolic effects of two fat compartments. Diabetes 2002;51:1005–15.ArticlePubMed
- 17. Cnop M, Havel PJ, Utzschneider KM, Carr DB, Sinha MK, Boyko EJ, et al. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: evidence for independent roles of age and sex. Diabetologia 2003;46:459–69.ArticlePubMedPDF
- 18. Bajaj M, Suraamornkul S, Hardies LJ, Pratipanawatr T, DeFronzo RA. Plasma resistin concentration, hepatic fat content, and hepatic and peripheral insulin resistance in pioglitazone-treated type II diabetic patients. Int J Obes Relat Metab Disord 2004;28:783–9.ArticlePubMedPDF
- 19. Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 2004;25:4–7.ArticlePubMed
- 20. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87–91.ArticlePubMed
- 21. Dandona P, Weinstock R, Thusu K, Abdel-Rahman E, Aljada A, Wadden T. Tumor necrosis factor-alpha in sera of obese patients: fall with weight loss. J Clin Endocrinol Metab 1998;83:2907–10.ArticlePubMed
- 22. Mohamed-Ali V, Goodrick S, Rawesh A, Katz DR, Miles JM, Yudkin JS, et al. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab 1997;82:4196–200.ArticlePubMed
- 23. Kubaszek A, Pihlajamaki J, Komarovski V, Lindi V, Lindstrom J, Eriksson J, et al. Promoter polymorphisms of the TNF-alpha (G-308A) and IL-6 (C-174G) genes predict the conversion from impaired glucose tolerance to type 2 diabetes: the Finnish Diabetes Prevention Study. Diabetes 2003;52:1872–6.ArticlePubMed
- 24. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004;306:457–61.ArticlePubMed
- 25. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004;114:1752–61.ArticlePubMedPMC
- 26. Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res 2006;45:42–72.ArticlePubMed
- 27. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006;116:1793–801.ArticlePubMedPMC
- 28. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, W et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 2005;11:191–8.ArticlePubMedPDF
- 29. Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature 2002;420:333–6.ArticlePubMedPDF
- 30. Dandona P, Aljada A, Mohanty P, Ghanim H, Hamouda W, Assian E, et al. Insulin inhibits intranuclear nuclear factor kappaB and stimulates IkappaB in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J Clin Endocrinol Metab 2001;86:3257–65.ArticlePubMed
- 31. Aljada A, Ghanim H, Mohanty P, Kapur N, Dandona P. Insulin inhibits the pro-inflammatory transcription factor early growth response gene-1 (Egr)-1 expression in mononuclear cells (MNC) and reduces plasma tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1) concentrations. J Clin Endocrinol Metab 2002;87:1419–22.ArticlePubMedPDF
- 32. Chen Z, Yu R, Xiong Y, Du F, Zhu S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis 2017;16:203. ArticlePubMedPMCPDF
- 33. Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 2013;10:330–44.ArticlePubMedPDF
- 34. Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 2001;50:1844–50.ArticlePubMed
- 35. Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, et al. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia 2005;48:634–42.ArticlePubMedPDF
- 36. Utzschneider KM, Kahn SE. Review: the role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab 2006;91:4753–61.ArticlePubMed
- 37. Seppala-Lindroos A, Vehkavaara S, Hakkinen AM, Goto T, Westerbacka J, Sovijarvi A, et al. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab 2002;87:3023–8.ArticlePubMed
- 38. Kelley DE, McKolanis TM, Hegazi RA, Kuller LH, Kalhan SC. Fatty liver in type 2 diabetes mellitus: relation to regional adiposity, fatty acids, and insulin resistance. Am J Physiol Endocrinol Metab 2003;285:E906–16.ArticlePubMed
- 39. Rhee EJ, Lee WY, Cho YK, Kim BI, Sung KC. Hyperinsulinemia and the development of nonalcoholic fatty liver disease in nondiabetic adults. Am J Med 2011;124:69–76.ArticlePubMed
- 40. Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014;59:713–23.ArticlePubMedPMC
- 41. Hudgins LC, Hellerstein M, Seidman C, Neese R, Diakun J, Hirsch J. Human fatty acid synthesis is stimulated by a eucaloric low fat, high carbohydrate diet. J Clin Invest 1996;97:2081–91.ArticlePubMedPMC
- 42. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343–51.ArticlePubMedPMC
- 43. Day CP, Saksena S. Non-alcoholic steatohepatitis: definitions and pathogenesis. J Gastroenterol Hepatol 2002;17 Suppl 3:S377–84.ArticlePubMedPDF
- 44. Roden M. Mechanisms of disease: hepatic steatosis in type 2 diabetes: pathogenesis and clinical relevance. Nat Clin Pract Endocrinol Metab 2006;2:335–48.ArticlePubMedPDF
- 45. Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 1997;99:846–54.ArticlePubMedPMC
- 46. Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem 2002;277:19353–7.ArticlePubMed
- 47. Tamura S, Shimomura I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J Clin Invest 2005;115:1139–42.ArticlePubMedPMC
- 48. Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 2004;432:1027–32.ArticlePubMedPDF
- 49. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab 2015;21:739–46.ArticlePubMed
- 50. Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007;45:1366–74.ArticlePubMed
- 51. Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2011;8:35–44.ArticlePubMedPDF
- 52. Gruben N, Shiri-Sverdlov R, Koonen DP, Hofker MH. Nonalcoholic fatty liver disease: a main driver of insulin resistance or a dangerous liaison? Biochim Biophys Acta 2014;1842:2329–43.ArticlePubMed
- 53. Lam TK, Carpentier A, Lewis GF, van de Werve G, Fantus IG, Giacca A. Mechanisms of the free fatty acid-induced increase in hepatic glucose production. Am J Physiol Endocrinol Metab 2003;284:E863–73.ArticlePubMed
- 54. Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, et al. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol 2004;18:2024–34.ArticlePubMed
- 55. Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem 2001;276:16683–9.ArticlePubMed
- 56. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006;116:3015–25.ArticlePubMedPMC
- 57. Knebel B, Haas J, Hartwig S, Jacob S, Kollmer C, Nitzgen U, et al. Liver-specific expression of transcriptionally active SREBP-1c is associated with fatty liver and increased visceral fat mass. PLoS One 2012;7:e31812.ArticlePubMedPMC
- 58. Ibdah JA, Perlegas P, Zhao Y, Angdisen J, Borgerink H, Shadoan MK, et al. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 2005;128:1381–90.ArticlePubMed
- 59. Otero YF, Stafford JM, McGuinness OP. Pathway-selective insulin resistance and metabolic disease: the importance of nutrient flux. J Biol Chem 2014;289:20462–9.ArticlePubMedPMC
- 60. Palma R, Pronio A, Romeo M, Scognamiglio F, Ventriglia L, Ormando VM, et al. The role of insulin resistance in fueling NAFLD pathogenesis: from molecular mechanisms to clinical implications. J Clin Med 2022;11:3649. ArticlePubMedPMC
- 61. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab 2008;7:496–507.ArticlePubMedPMC
- 62. Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014;59:2034–42.ArticlePubMed
- 63. Sorrentino P, Terracciano L, D'Angelo S, Ferbo U, Bracigliano A, Vecchione R. Predicting fibrosis worsening in obese patients with NASH through parenchymal fibronectin, HOMA-IR, and hypertension. Am J Gastroenterol 2010;105:336–44.ArticlePubMedPDF
- 64. Fujii H, Imajo K, Yoneda M, Nakahara T, Hyogo H, Takahashi H, et al. HOMA-IR: an independent predictor of advanced liver fibrosis in nondiabetic non-alcoholic fatty liver disease. J Gastroenterol Hepatol 2019;34:1390–5.ArticlePubMedPDF
- 65. Aller R, Siguenza R, Pina M, Laserna C, Antolin B, Burgueno B, et al. Insulin resistance is related with liver fibrosis in type 2 diabetic patients with non-alcoholic fatty liver disease proven biopsy and Mediterranean diet pattern as a protective factor. Endocrine 2020;68:557–63.ArticlePubMedPDF
- 66. Koo DJ, Lee MY, Jung I, Moon SJ, Kwon H, Park SE, et al. Baseline homeostasis model assessment of insulin resistance associated with fibrosis progression in patients with nonalcoholic fatty liver disease without diabetes: a cohort study. PLoS One 2021;16:e0255535.ArticlePubMedPMC
- 67. Koo DJ, Lee MY, Jung I, Moon SJ, Kwon H, Park SE, et al. Changes in insulin resistance index and the risk of liver fibrosis in patients with nonalcoholic fatty liver disease without diabetes: Kangbuk Samsung Health Study. Endocrinol Metab (Seoul) 2021;36:1016–28.ArticlePubMedPMCPDF
- 68. Long MT, Noureddin M, Lim JK. AGA Clinical Practice Update: diagnosis and management of nonalcoholic fatty liver disease in lean individuals: expert review. Gastroenterology 2022;163:764–74.ArticlePubMed
- 69. Musso G, Cassader M, Rosina F, Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia 2012;55:885–904.ArticlePubMedPDF
- 70. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015;149:367–78.ArticlePubMed
- 71. Sinn DH, Kang D, Cho SJ, Paik SW, Guallar E, Cho J, et al. Weight change and resolution of fatty liver in normal weight individuals with nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol 2021;33(1S Suppl 1):e529–34.Article
- 72. Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005;54:603–8.ArticlePubMedPDF
- 73. Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010;51:121–9.ArticlePubMedPMC
- 74. Kirk E, Reeds DN, Finck BN, Mayurranjan SM, Patterson BW, Klein S. Dietary fat and carbohydrates differentially alter insulin sensitivity during caloric restriction. Gastroenterology 2009;136:1552–60.ArticlePubMedPMC
- 75. Haufe S, Engeli S, Kast P, Bohnke J, Utz W, Haas V, et al. Randomized comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology 2011;53:1504–14.ArticlePubMed
- 76. St George A, Bauman A, Johnston A, Farrell G, Chey T, George J. Independent effects of physical activity in patients with nonalcoholic fatty liver disease. Hepatology 2009;50:68–76.ArticlePubMed
- 77. Sung KC, Ryu S, Lee JY, Kim JY, Wild SH, Byrne CD. Effect of exercise on the development of new fatty liver and the resolution of existing fatty liver. J Hepatol 2016;65:791–7.ArticlePubMed
- 78. Kwak MS, Kim D, Chung GE, Kim W, Kim YJ, Yoon JH. Role of physical activity in nonalcoholic fatty liver disease in terms of visceral obesity and insulin resistance. Liver Int 2015;35:944–52.ArticlePubMedPDF
- 79. Wong VW, Wong GL, Chan RS, Shu SS, Cheung BH, Li LS, et al. Beneficial effects of lifestyle intervention in non-obese patients with non-alcoholic fatty liver disease. J Hepatol 2018;69:1349–56.ArticlePubMed
- 80. Marchesini G, Brizi M, Bianchi G, Tomassetti S, Zoli M, Melchionda N. Metformin in non-alcoholic steatohepatitis. Lancet 2001;358:893–4.ArticlePubMed
- 81. Bugianesi E, Gentilcore E, Manini R, Natale S, Vanni E, Villanova N, et al. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol 2005;100:1082–90.ArticlePubMed
- 82. Li Y, Liu L, Wang B, Wang J, Chen D. Metformin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Biomed Rep 2013;1:57–64.ArticlePubMedPMC
- 83. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med 2016;165:305–15.ArticlePubMed
- 84. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362:1675–85.ArticlePubMedPMC
- 85. Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008;135:1176–84.ArticlePubMed
- 86. Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 2006;355:2297–307.ArticlePubMed
- 87. Lee YH, Kim JH, Kim SR, Jin HY, Rhee EJ, Cho YM, et al. Lobeglitazone, a novel thiazolidinedione, improves non-alcoholic fatty liver disease in type 2 diabetes: its efficacy and predictive factors related to responsiveness. J Korean Med Sci 2017;32:60–9.ArticlePubMedPMCPDF
- 88. Tuccori M, Filion KB, Yin H, Yu OH, Platt RW, Azoulay L. Pioglitazone use and risk of bladder cancer: population based cohort study. BMJ 2016;352:i1541. ArticlePubMedPMC
- 89. Lewis JD, Habel LA, Quesenberry CP, Strom BL, Peng T, Hedderson MM, et al. Pioglitazone use and risk of bladder cancer and other common cancers in persons with diabetes. JAMA 2015;314:265–77.ArticlePubMed
- 90. Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA 2011;305:1659–68.ArticlePubMedPMC
- 91. Vadarlis A, Antza C, Bakaloudi DR, Doundoulakis I, Kalopitas G, Samara M, et al. Systematic review with meta-analysis: the effect of vitamin E supplementation in adult patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol 2021;36:311–9.ArticlePubMedPDF
- 92. Said A, Akhter A. Meta-analysis of randomized controlled trials of pharmacologic agents in non-alcoholic steatohepatitis. Ann Hepatol 2017;16:538–47.ArticlePubMed
- 93. Miller ER 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 2005;142:37–46.ArticlePubMed
- 94. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016;387:679–90.ArticlePubMed
- 95. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med 2021;384:1113–24.ArticlePubMed
- 96. Kahl S, Gancheva S, Strassburger K, Herder C, Machann J, Katsuyama H, et al. Empagliflozin effectively lowers liver fat content in well-controlled type 2 diabetes: a randomized, double-blind, phase 4, placebo-controlled trial. Diabetes Care 2020;43:298–305.ArticlePubMedPDF
- 97. Phrueksotsai S, Pinyopornpanish K, Euathrongchit J, Leerapun A, Phrommintikul A, Buranapin S, et al. The effects of dapagliflozin on hepatic and visceral fat in type 2 diabetes patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol 2021;36:2952–9.ArticlePubMedPDF
- 98. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016;150:1147–59.ArticlePubMed
- 99. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013;145:574–82.ArticlePubMed
- 100. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 2015;385:956–65.ArticlePubMed
Citations
Citations to this article as recorded by

 , Won-Young Lee2
, Won-Young Lee2

 PubReader
PubReader ePub Link
ePub Link Cite
Cite