ABSTRACT
- There are various types of adverse reactions to antiarrhythmic drugs (AADs). Proarrhythmia, which refers to an exacerbation of the preexisting arrhythmia or occurrence of a new arrhythmia, may occur under the therapeutic concentration of an AAD. Bradyarrhythmia is the most common type of proarrhythmia due to AADs, and prior myocardial infarction and old age are known risk factors. Atrial flutter with 1:1 atrioventricular conduction usually occurs during rhythm control of atrial fibrillation with class IC AADs. QT prolongation due to AADs, mainly class III AADs, elevates the risk of torsade de pointes by triggered activity due to early afterdepolarization. The addition of clinical factors that promote QT prolongation, such as hypokalemia, hypomagnesemia, female sex, and bradycardia, increases the risk of developing torsade de pointes. Proarrhythmic monomorphic ventricular tachycardia usually occurs as a result of slow conduction and disparity of refractoriness due to class IC AADs. In patients with preexisting left ventricular systolic dysfunction or structural heart disease, the risk of hypotension or cardiogenic shock caused by negative inotropic effects due to AADs should be considered. To prevent these major adverse reactions to AADs, we need to understand the electrophysiologic properties of AADs in detail. Furthermore, the risk of proarrhythmia could be heightened by interplay with clinical factors, such as electrolyte unbalances, heart rate, and hepatic/renal or myocardial dysfunction. Sufficient awareness about drug-drug interactions, which may affect the metabolism of AADs, will improve patient safety during the management of arrhythmia.
-
Keywords: Anti-arrhythmia agents; Proarrhythmia; Drug interactions
INTRODUCTION
- Antiarrhythmic drugs (AADs) are an important therapeutic modality for rhythm control in patients with cardiac arrhythmias. In particular, atrial fibrillation (AF) has emerged as the most important arrhythmic disorder of the present era, as Korea is rapidly becoming an aging society. Because early rhythm control has shown favorable clinical outcomes compared with rate control in recent trials [1,2], the use of AADs is increasing rapidly. However, the clinical outcomes of management with AADs are not all roses. All AADs, like all other medications, can cause several adverse reactions. It has a broad spectrum from self-limiting, mild adverse reactions to severe, fatal adverse reactions, including fatal arrhythmias. The definition of an adverse drug reaction is a significantly harmful or unpleasant response resulting from treatment with medication [3]. AADs are like double-edged swords—there is not only an antiarrhythmic effect, but also a proarrhythmic risk. Since proarrhythmia is often very fatal and is considered the most important category of adverse reactions to AADs, we focus on proarrhythmia in this paper. We also introduce major drug-drug interactions that affect the serum concentration of AADs and elevate the risk of adverse reactions to AADs.
CLASSIFICATION OF ANTIARRHYTHMIC DRUGS
- The Vaughan-Williams classification remains the most popular and familiar method of classifying AADs [4]. It has recently been expanded to include potential AADs [5], and class I to IV AADs remain the cornerstone of the management of arrhythmic disorders. Another approach toward a new classification of AADs, called the Sicilian Gambit, increased the level of understanding of the mechanism of AADs and parameters associated with vulnerability to arrhythmias; however, it failed to lead to widespread acceptance and clinical use due to its complexity [6]. Each AAD has an additional mechanism of pharmacological action beyond its known major mode of action (Table 1) [6,7]. Furthermore, the interplay between AADs and clinical environments leads to a difference in the degree of pharmacologic actions. Using AADs without a detailed understanding of these complexities may increase the occurrence of proarrhythmic adverse reactions.
PROARRHYTHMIA
- Definition
- Proarrhythmia is defined as a progression of preexisting arrhythmia or the occurrence of a new arrhythmia resulting from therapy for arrhythmic control with medication of a clinically usually therapeutic, nontoxic concentration [8].
- Antiarrhythmic drug-induced specific arrhythmias
Bradyarrhythmia
- Bradyarrhythmia is known as the most common manifestation of proarrhythmia resulting from the use of AADs [9]. Bradyarrhythmia could occur due to suppression of automaticity of the sinoatrial node or conduction ability via the atrioventricular (AV) node and the His-Purkinje system. The incidence of bradyarrhythmia due to AADs is unknown. Previous myocardial infarction and old age have been suggested as risk factors for bradyarrhythmia in patients who have undergone rhythm control for AF [9]. The incidence of AF is much higher in older people, and physicians frequently prescribe AADs for rhythm management in symptomatic paroxysmal AF patients with old age. Even in the absence of evidence of preexisting conduction system dysfunction, we often meet patients who complain of faintness or syncope after treatment with AADs, which are suspected to be symptoms due to sick sinus syndrome or post-tachycardia pause. Therefore, AADs should be started at a low dose combined with periodic follow-up, especially in older patients. Cardiac death due to bradyarrhythmia is rare; however, pause-dependent torsade de pointes could occur, and traumatic hemorrhage due to loss of consciousness is another fatal complication.
Atrial flutter with 1:1 atrioventricular conduction
- Atrial flutter with 1:1 AV conduction can sometimes be observed when we try rhythm control therapy in patients with AF. Class IC AADs are used frequently in patients without structural heart disease to treat AF or supraventricular tachycardia. Its known incidence rate during AF treatment with flecainide or propafenone is about 3.5% to 5.0% [7]. Class I AADs inhibit phase 0 depolarization by blocking sodium channels. These drugs make the conduction velocity slow in the heart. When a class I agent was used to treat atrial flutter, the flutter cycle length could be slowed, and this could occur in patients with AF as the AF organized to the atrial flutter. AV conduction via the AV node could be accelerated to 1:1 AV conduction due to the prolongation of the atrial flutter cycle length. In particular, class IC AADs bind to and dissociate from the sodium channel more slowly than other class I AADs [10]. Therefore, use-dependency is a pharmacologic characteristic of class IC AADs, and the acceleration of AV conduction might occur more easily than in other class I AADs. Furthermore, the sodium channel blocking effect could also delay ventricular conduction via the bundle branch and muscle-to-muscle conduction. According to an animal study, conduction delay due to flecainide is more remarkably presented in the ventricular myocardium than in the Purkinje fibers [11]. This aberrant and delayed intraventricular conduction makes the QRS complex wide and bizarre, frequently resulting in a pattern that is difficult to distinguish from true ventricular tachycardia. According to a recent report, the initial deflection of the QRS complex in lead V6 could be helpful for distinguishing between these arrhythmias [12]. Fig. 1 shows an example of atrial flutter with 1:1 AV conduction, which occurred during rhythm control with flecainide in a patient with paroxysmal AF. Due to this proarrhythmic risk, an AV nodal blocking agent should be used when we try to use AADs for rhythm control in cases of AF or atrial flutter.
Sustained monomorphic ventricular tachycardia
- Sustained monomorphic ventricular tachycardia (VT) can occur as a complication of any type of AAD. However, the conduction-blocking effect due to class IC AADs is the most common pathophysiology of proarrhythmic monomorphic VT. The known predictors of sustained monomorphic VT are left ventricular systolic dysfunction and a history of undergoing rhythm control for ventricular tachyarrhythmia [13,14]. However, Falk [15] have reported sustained VT cases related to flecainide use for rhythm control in patients with AF and normal left ventricular function. Josephson [16] suggested that this phenomenon potentially was reentrant VT due to slow conduction and a disparity of refractoriness in flecainide use. Of particular note, this use-dependent depression of conduction associated with a shortening of refractoriness could progress as a result of exercise or catecholamine use. As a result, sustained monomorphic VT could be more easily induced in those situations. The feature of proarrhythmic monomorphic VT due to flecainide tends to appear in the form of a wide sine wave. Fig. 2 shows an example of sustained monomorphic VT during treatment with flecainide, which occurred in a patient without evidence of structural heart disease. This ventricular flutter-like tachyarrhythmia could be fatal and not be converted to sinus rhythm by electrical cardioversion [16]. This proarrhythmic effect could be caused by a too rapid escalation in the AAD dose.
Torsade de pointes
- Torsade de pointes (TdP) is a very well-known life-threatening ventricular tachyarrhythmia. This is polymorphic VT occurring at the prolonged QT interval, which has characteristics of the QRS complex’s morphology with alternating electric polarity and amplitude, and the peak of the QRS complex appears to be twisting around the isoelectric line. TdP is usually short-lived; however, multiple episodes of TdP could degenerate into ventricular fibrillation [17]. Among the AADs, class IA or Ⅲ drugs, which block outward potassium currents during the phase 3 period of the action potential, are commonly associated with TdP. These agents lead to the prolongation of the action potential duration, especially the repolarization period [18], and the QT prolongation is markedly aggravated at the following beat immediately after a post-ectopic pause or a long R-R interval [19]. This results in intracellular calcium overloading due to the prolonged plateau phase, and augmented calcium influx might have a crucial role in the initial mechanism of TdP, which is activity triggered by early afterdepolarization [20]. When other factors, such as hypokalemia, hypomagnesemia, female sex, bradycardia, and renal failure, are superimposed on the QT prolongation due to AADs, the risk of TdP is increased [20,21]. Therefore, we usually encounter TdP cases in patients with chronic wasting conditions who have undergone treatment in the intensive care unit, with the frequent use of diuretics and AADs for AF.
- Interestingly, the risk of TdP in patients who take amiodarone is relatively low despite the degree of QT prolongation (Table 2) [8]. However, for sotalol, the incidence of TdP is more than 5% at doses of more than 320 mg/day [18]. This means that the mechanism of the prolongation of the repolarization period might be more important than the degree of the prolongation of the QTc interval. Although it is still unclear, the low risk of TdP in amiodarone might be explained by its blocking effects on multiple ionic currents and unremarkable increment of QT dispersion [22,23]. Furthermore, reverse use-dependence is characteristic of sotalol, in contrast to amiodarone. The β-blocking effect combined with the reverse use-dependence of sotalol can provoke marked QT prolongation in the diastolic period, especially during bradycardia, and elevate the risk of TdP. In contrast, prolongation of the action potential by amiodarone at normal and fast heart rates is similar because its action is less time-dependent and voltage-dependent; that is, reverse use-dependence is not characteristic of amiodarone [24]. Self-terminating TdP with recurrent syncope in patients who take quinidine for the management of AF was first reported by Selzer and Wray [25]. This is also named “quinidine syncope” and usually occurs within 1 to 3 hours after the last dose. Quinidine syncope could be life-threatening and is associated with significant QT prolongation. The QT prolongation could be aggravated due to the reverse use-dependence of its outward potassium current inhibition [24]. Meanwhile, TdP may occur due to an idiosyncratic reaction at low dosages and concentrations of quinidine [18]. Dronedarone also causes prolongation of the QT interval as a class III AAD; however, the degree of QT prolongation is modest at a daily dose of 800 mg [26], and reported TdP cases are extremely rare according to the results of clinical trials [27,28].
Ventricular fibrillation
- Ventricular fibrillation (VF) during rhythm management with AADs was evaluated intensively in the 1980s [29,30]. Left ventricular systolic dysfunction was identified as a risk factor for AAD-associated VF [30]. In those studies, the AADs that provoked VF were class IA agents, such as procainamide, quinidine, and disopyramide. In the present clinical practice of the management of tachyarrhythmia, the clinical indication of these class IA AADs is very narrow, and these AADs are not available in clinical practice in Korea, except for quinidine.
- Sodium channel blockers, including class IC, ajmaline, and procainamide can induce Brugada-pattern electrocardiography (ECG) in patients with normal pretreatment ECG. A drug-induced Brugada pattern may be observed more frequently during the treadmill test, and the patients with this change are usually asymptomatic. However, VF and sudden cardiac death could still occur, so the offending drug should be discontinued [31].
Interplay with extraneous factors
- The risk of proarrhythmic effects of AADs could be heightened in certain clinical environments. The serum concentration of AADs varies depending on drug metabolism. Therefore, the renal and/or hepatic excretion of a specific medication or numerous drug-drug interactions could elevate the risk of proarrhythmia [8]. Sotalol should be prescribed cautiously in patients with renal insufficiency, and the use of lidocaine in the presence of severe hepatic dysfunction may elevate the risk. The details of drug-drug interactions will be discussed below. The concomitant presence of hypokalemia or hypomagnesemia elevates the risk of TdP. Hyperkalemia decreases conduction velocity and promotes bradycardia due to dysfunction of the sinoatrial node and/or AV node. During a rapid heart rate, the pharmacological effect of class IC AADs is exaggerated due to their unique pharmacokinetic feature, which is slow binding to and dissociation from the sodium channel [32]. In contrast, reverse use-dependency is characteristic of class IA AADs and sotalol; therefore, the use of these agents should be avoided in patients with advanced bradycardia to reduce the risk of QT prolongation and TdP [24]. Additionally, all class I AADs should be avoided in patients with myocardial ischemia to reduce mortality due to arrhythmia [33].
NEGATIVE INOTROPIC EFFECTS
- Most AADs have a negative inotropic effect; however, whether a clinically significant negative inotropic effect is overt may vary depending on multiple variables, such as underlying structural heart disease or myocardial function and the loading conditions of the AAD [34,35]. Therefore, the hemodynamic result of a given AAD could be different according to the tested conditions. Besides, the evidence is relatively weak to discuss the negative inotropic effect of AADs in detail and the results of animal studies are not always correlated to the results of clinical studies. That is, it is difficult to summarize the negative inotropic effect of AADs.
- Class IC AADs, flecainide and propafenone, which have a strong sodium channel-blocking effect, decrease cardiac contractility. It is also known that propafenone has a β-blocking effect. This is related to the alteration of sodium/calcium inter-dependence and a decrease in intracellular calcium [7]. Class IC AADs should not be used in the presence of structural heart disease, especially ischemic cardiomyopathy [33].
- Sotalol inhibits rapid delayed rectifier potassium current (Ikr). Sotalol could aggravate heart failure. According to a prior study that evaluated 3,257 patients treated for arrhythmias, heart failure occurred in 3.3% of patients during a median of 156 days of follow-up [36]. Heart failure was more prevalent in patients with a prior history of heart failure, cardiomyopathy, or structural heart disease.
- Amiodarone has a minimal effect on the left ventricular ejection fraction (LVEF) in patients with near-normal or normal LVEF [34]. Rather, a study showed that the LVEF slightly increased due to reduced afterload by a vasodilatory effect [37]. Amiodarone is widely used as the safest possible AAD in patients with structural heart disease and heart failure with and without atrial or ventricular tachyarrhythmia. However, caution is necessary because slight reductions of the LVEF could occur in patients with systolic dysfunction or structural heart disease, especially when intravenous (IV) amiodarone is infused at a high dose [38]. Several case reports have described cardiogenic shock after an IV bolus injection in patients with left ventricular systolic dysfunction [39,40]. Hypotension caused by IV amiodarone may be partially explained by the cosolvents (polysorbate 80 or benzyl alcohol), used when amiodarone is prepared in an IV formulation [41].
- Several studies that enrolled populations including patients with heart failure evaluated the clinical outcomes of dronedarone [42,43]. These studies showed that dronedarone increased the rate of heart failure and cardiovascular death. Therefore, dronedarone should not be used in patients with unstable conditions, with prior or current heart failure or systolic dysfunction [7]. Although the electrophysiological mechanism of dronedarone is similar to that of amiodarone, the difference in mortality in patients with heart failure seems to be explained only by a reduction in free intracellular calcium and amplitude of contraction, which was demonstrated in an experiment with a guinea pig ventricular cell model, because the experimental evidence is limited [44].
DRUG INTERACTIONS AND ADVERSE EFFECTS OF SPECIFIC ANTIARRHYTHMIC DRUGS
- Herein, we focus on the AADs that are available in present clinical practice in Korea. Table 3 summarizes the adverse effects of major AADs and the precautions required to minimize the adverse reactions during treatment.
- Flecainide
- Because flecainide is mainly metabolized with hepatic hydroxylation via cytochrome P450 (CYP) 2D6 isoenzyme, we should consider the risk of adverse reactions due to over-concentration with concomitant administration of other medications that could modulate the CYP isoenzyme. Several selective serotonin reuptake inhibitors, including paroxetine and fluoxetine, can increase the plasma concentration of flecainide by CYP2D6 inhibition [45]. Among the various β-blockers, the concomitant use of propranolol with flecainide could elicit synergistic hypotension, which was not reported with other β-blockers [46]. The concomitant use of amiodarone increases the mean dose-adjusted plasma level of flecainide by up to 50% [47]. Serum digoxin concentration increased by 24% when flecainide is coadministered [46]. Flecainide is excreted unchanged in urine at about 35%, and dose reduction should be applied in patients with a glomerular filtration rate (GFR) <35 mL/min/1.73m2 [7]. Infrequently, neutropenia, hepatotoxicity, and impotence could occur [48–50].
- Propafenone
- Extensive first-pass metabolism by CYP2D6 is the pharmacodynamic characteristic of propafenone. When warfarin is administered with propafenone, the warfarin concentration is elevated by 38% due to an unknown mechanism; therefore, an approximately 33% dose reduction of warfarin and follow-up of the international normalized ratio should be applied [51]. Serum digoxin concentration increases by 60% when propafenone is coadministered [52]. Hepatotoxicity and lupus-like syndromes are reported as potential adverse reactions to this AAD [50,53–55].
- Amiodarone
- Amiodarone has a remarkably long half-life of 55 days [56]. Amiodarone is usually metabolized by CYP3A4, and it is a potent inhibitor of many CYP enzymes, including CYP2C9 and CYP2D6 [57]. Therefore, drug-drug interactions should be considered whenever concomitant medical treatment is administered. Inhibition of CYP2C9 increased the warfarin concentration [58]. A reduced dose of warfarin according to the dose of amiodarone should be applied, and periodic follow-up of the international normalized ratio needs to be performed [59]. Cyclosporine coadministration leads to an increase in the serum cyclosporine concentration by inhibiting CYP3A4; therefore, dose reduction of cyclosporine is needed, by about 50%, and the drug concentration should be regularly monitored [60]. Amiodarone inhibits the metabolism of several statins, including simvastatin and lovastatin, via the inhibition of CYP3A4, increasing the likelihood of statin toxicity [57].
- When we prescribe amiodarone to patients with arrhythmia, attention should be paid to the risk of adverse effects related to the AAD. Most of the adverse effects related to amiodarone are known to be dose related. The 1-year risk of pulmonary toxicity due to amiodarone is reported as about 1% [61]. Furthermore, 5% to 10% of them manifest as fatal situations [62]. The underlying mechanisms of pulmonary toxicity were reported as results of direct cellular toxicity, phospholipidosis, immune reactions, and oxidative stress. Fig. 3 shows a case of a patient with amiodarone-induced pulmonary toxicity. Hyperthyroidism and hypothyroidism could all occur due to amiodarone. The onset is not usually dose related, and most patients remain euthyroid. If clinically indicated, a continuation of amiodarone combined with hormonal replacement or antihyperthyroid management with methimazole or propylthiouracil would be considered. Photoallergic and phototoxic skin reactions could occur related to amiodarone; these are explained by its strong lipophilicity and affinity to the cell membrane. Therefore, patients should be informed of the need to apply sunblock. Blurred vision due to corneal microdeposits, known as vortex keratopathy, is usually a reversible adverse effect. A transient elevation of serum aminotransferase could be observed in about 25% of patients [63]. Amiodarone-related liver injury is considered to be an idiosyncratic phenomenon due to direct hepatotoxicity, and it is usually benign and asymptomatic. According to the post hoc data from the DIONYSOS trial, the frequency of liver enzyme elevation was comparable between amiodarone and dronedarone [64]. Therefore, routine monitoring of liver toxicity is required. Central nervous system symptoms, such as tremors, ataxia, insomnia, and/or vivid dream, and peripheral neuropathic symptoms could occur. The Heart Rhythm Society recommends the serial monitoring of amiodarone-related adverse reactions as Table 4 [65]. High-resolution computed tomography of the lung is recommended if the clinical suspicion of pulmonary toxicity is present, such as unexplained cough or dyspnea, especially in patients with underlying lung disease [65].
- Dronedarone
- Dronedarone, a benzofuran derivative, is a chemical analog of amiodarone without iodine-related adverse effects. However, it is still lipophilic and is metabolized by CYP3A4 like amiodarone. When combining moderate CYP3A4 inhibitors, such as verapamil and diltiazem, the dronedarone concentration is increased by 1.4-fold to 1.7-fold [66]. Potent CYP3A4 inhibitors, such as antifungals and macrolide antibiotics, should not be coadministered with dronedarone because of the risk of adverse effects due to elevation of serum concentrations [67]. Moreover, since dronedarone is a P-glycoprotein (P-gp) inhibitor, it can increase the concentration of digoxin and dabigatran during concomitant treatment [67]. Reduction of the digoxin dose by half is recommended to avoid adverse effects [68]. A reduced dabigatran dose of 75 mg twice daily should be administered in patients with creatine clearance (CrCl) between 30 and 50 mL/min, and coadministration of dabigatran should be avoided if CrCl is less than 30 mL/min [69]. The dose of edoxaban, which is a strong P-gp inhibitor, is recommended to reduce by 50% when edoxaban is used with dronedarone [70]. Because rivaroxaban induces the inhibition of P-gp and CYP3A4, considerable interactions with dronedarone could be expected. Although there are limited safety data, the combined use of rivaroxaban should be avoided if the CrCl is less than 80 mL/min [71,72].
- Physicians sometimes encounter patients complaining of gastrointestinal adverse reactions, including abdominal pain, dyspepsia, diarrhea, nausea, and vomiting, during treatment with dronedarone. Fatigue and asthenia are also reported adverse effects [64]. Although there was a report of acute liver failure in dronedarone-prescribed patients in 2011, subsequent data consistently suggested that the risk of hepatic injury associated with dronedarone is lower or comparable to the risk of other AADs [64].
- Sotalol
- Sotalol is a racemic mixture of the l-isomer, with β-blocking activity and class III activity, and the d-isomer, which only exerts class III activity, inhibiting Ikr [73]. This AAD has much simpler pharmacokinetics than amiodarone due to its predominantly renal metabolism; therefore, it is relatively free from drug-drug interactions via the hepatic CYP coenzyme. This hydrophilic β-blocker is excreted unchanged in the urine and should be avoided in patients undergoing hemodialysis. Dose reduction to 50% is recommended in patients with chronic renal insufficiency and 25% in those with severe renal disease (GFR <30 mL/min/1.73m2) to prevent proarrhythmic events [7]. Sotalol has a β-blocking effect with a potency of about one-third that of propranolol [74]. Its known adverse effects are usually similar to the adverse reactions related to other β-blockers, such as fatigue, dizziness, and dyspnea, without evidence of organ toxicity.
- Lidocaine
- Hepatic metabolism via CYP1A2 is the main process of lidocaine elimination [75]. Conditions that decrease hepatic blood flow include cardiogenic shock and severe liver disease, and concomitant treatment with CYP3A4 inhibitors, such as erythromycin, could elevate the lidocaine levels and exacerbate the risk of adverse reactions [76]. Central nervous system toxicity is the major adverse effect of IV lidocaine. Tremor, dysarthria, lightheadedness, agitation, hallucinations, and personality change can occur. These adverse effects are usually dose-dependent and resolve after discontinuation of the agent [77].
- Quinidine
- Quinidine has a relatively narrow clinical indication as an AAD in recent clinical practice and is usually prescribed in patients with Brugada syndrome, short QT syndrome, or idiopathic VF. Quinidine is mainly metabolized by CYP3A4 [78]. The coadministration of itraconazole can significantly elevate the serum quinidine concentration [79]. Quinidine can increase the concentration of propranolol via the suppression of CYP2D6; therefore, reducing the dose of propranolol should be considered to reduce the risk of augmented beta-blocking effects during concomitant treatment [80]. Gastrointestinal symptoms, including diarrhea, are the most common adverse reaction. Tinnitus, hearing loss, dizziness, visual disturbance, and psychosis, also known as cinchonism, could occur. Immune-mediated fever, hemolytic anemia, thrombocytopenia, and leukopenia have also been reported [81].
CONCLUSIONS
- Most AADs could have multiple effects on channels, receptors, and pumps and also alter the hemodynamics and autonomic nervous system. The multiple electrophysiologic actions of AADs interacting with the variable underlying substrates present in each patient determine whether the clinical effect will be proarrhythmic or antiarrhythmic. Reducing adverse reactions to AADs requires an in-depth understanding of AADs, including their electrophysiological properties and drug-drug interactions with concomitantly used drugs, as well as careful monitoring.
ARTICLE INFORMATION
-
Ethical statements
Not applicable.
-
Conflicts of interest
The author has no conflicts of interest to declare.
-
Funding
None.
Fig. 1.Atrial flutter with 1:1 atrioventricular conduction. (A) This 51-year-old male patient was transferred due to wide QRS complex tachycardia. He had been treated with flecainide and verapamil due to paroxysmal atrial fibrillation. (B) The tachycardia was not terminated by adenosine, and the rhythm postadenosine infusion was compatible with typical atrial flutter. Subsequently during catheter ablation for his atrial fibrillation, atypical atrial flutter was induced by burst pacing under intravenous flecainide infusion. (C) The QRS complex morphology of the induced atrial flutter was the same as that of the initial electrocardiography of wide QRS tachycardia. This could be accepted as a finding providing further support that (A) the QRS widening of electrocardiography occurred by aberrant conduction, affected by flecainide.
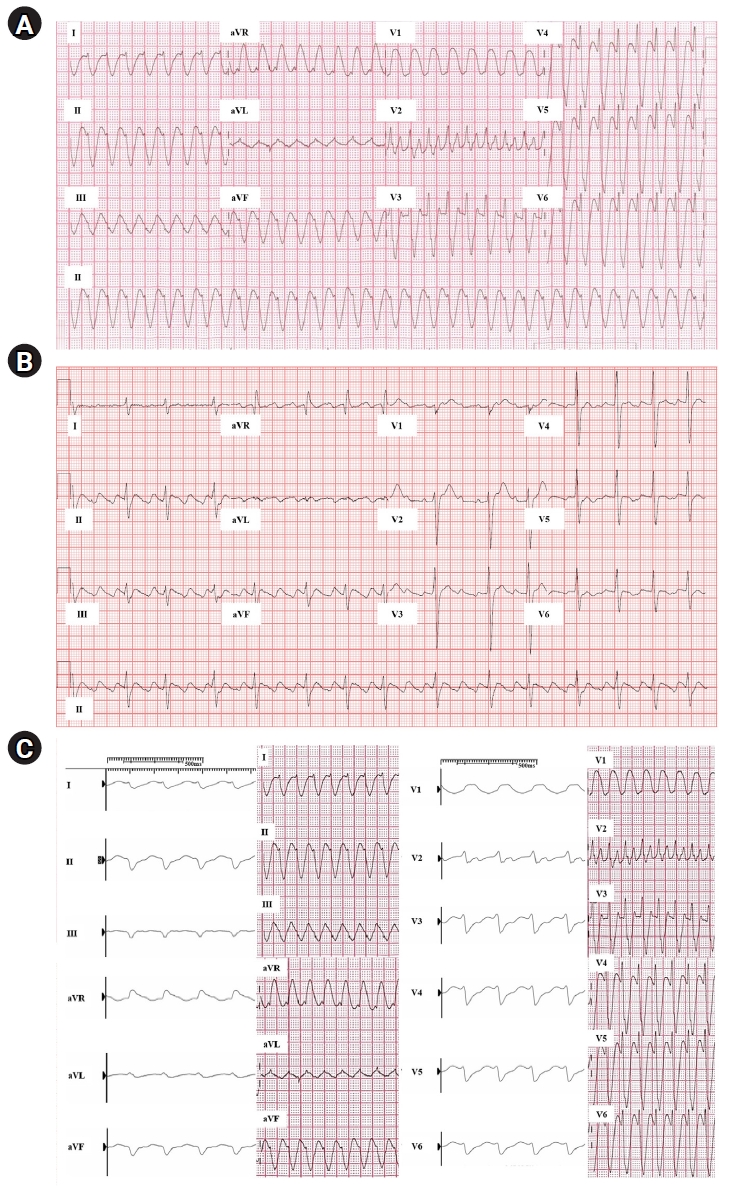
Fig. 2.Monomorphic ventricular tachycardia. This 44-year-old female patient underwent cavotricuspid isthmus ablation and implantation of a permanent pacemaker (DDDR mode) due to typical atrial flutter and sick sinus syndrome. She had taken flecainide (50 mg twice daily), and diltiazem (30 mg twice daily) due to paroxysmal atrial fibrillation. She visited the emergency room due to palpitation. Her blood pressure was 90/60 mmHg. (A) Initial electrocardiography showed wide QRS tachycardia of 180 beats/min. The tachycardia was not responsive to intravenous adenosine, and electrical cardioversion was performed for sinus conversion. (B) Interrogation of the pacemaker showed that the clinical tachycardia was compatible with ventricular tachycardia. The morphology of the QRS complex during tachycardia resembles a sine wave.
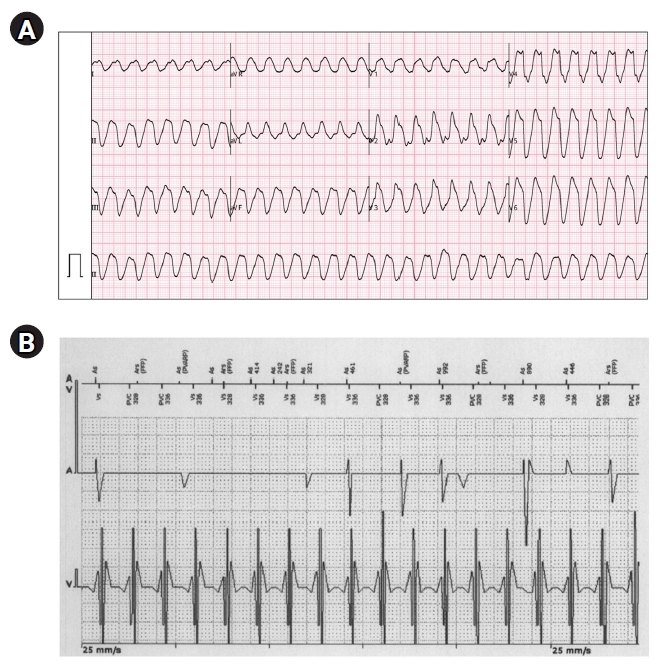
Fig. 3.Amiodarone-induced pulmonary toxicity. (A) Chest radiography and (B) a high-resolution computed tomography of a 58-year-old male patient with amiodarone-induced pulmonary toxicity. This patient with persistent atrial fibrillation had been treated with oral amiodarone. (A) Chest radiography was performed due to mild dyspnea during exertion, and increased opacity in the right middle and lower lung fields was observed. (B) Diffuse peribronchial consolidations with ground-glass opacity and nodular density at both lung fields were detected on computed tomography.
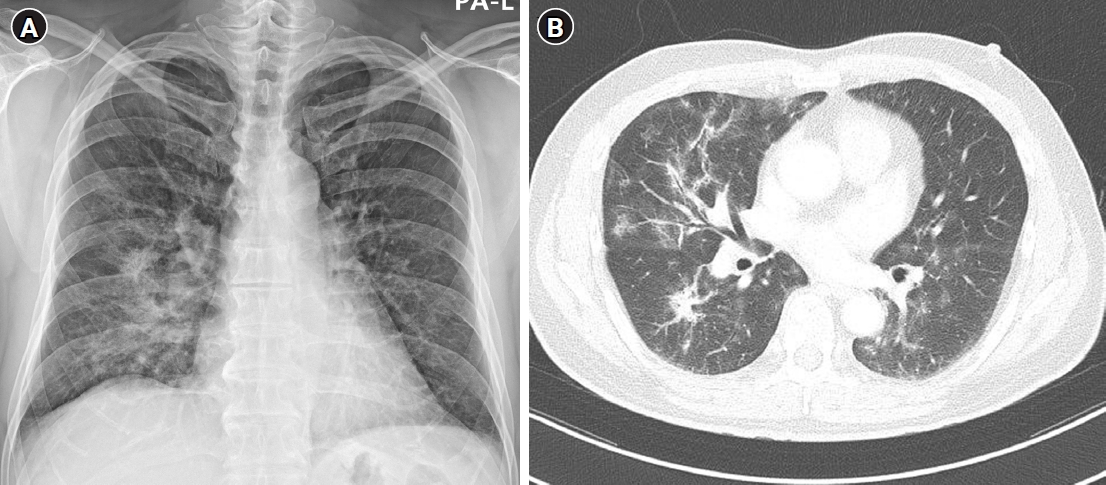
Table 2.Proarrhythmic effects of antiarrhythmic drugs
|
Variable |
TdP |
VFa)
|
VTa)
|
AFL 1:1 AV conduction |
Bradyarrhythmia |
|
Procainamide |
1%–2% |
++ |
+ |
+ |
+ |
|
Quinidine |
0.02 |
++ |
+ |
++ |
|
|
Lidocaine |
|
Rare |
Rare |
Rare |
Rare |
|
Mexiletine |
|
Rare |
Rare |
|
Rare |
|
Propafenone |
Rare |
+ |
++ |
+++ |
++ |
|
Flecainide |
Rare |
+ |
++ |
+++ |
++ |
|
Sotalol |
2–5% |
+ |
+ |
+ |
+++ |
|
Amiodarone |
<1% |
+ |
+ |
+ |
+++ |
Table 3.Summary of adverse effects of major antiarrhythmic drugs and precautions during treatment
|
Drug |
Adverse effect |
Precaution |
|
Flecainide |
AFL with 1:1 AV conduction, bradyarrhythmia, sustained monomorphic VT, conduction velocity slowing, widening of QRS complex, negative inotropic effect, hepatotoxicity |
Contraindication (structural heart disease, ischemic heart disease), DDI with CYP2D6 inhibitors (SSRI, amiodarone), synergistic hypotension combined with propranolol, dose reduction during combined with digoxin or amiodarone, and when GFR <35 mL/min/1.73m2
|
|
Propafenone |
AFL with 1:1 AV conduction, bradyarrhythmia, sustained monomorphic VT, conduction velocity slowing, widening of QRS complex, negative inotropic effect, hepatotoxicity |
Contraindication (structural heart disease, ischemic heart disease), dose reduction during combination with warfarin or digoxin |
|
Amiodarone |
QT prolongation, TdP, bradyarrhythmia, extracardiac toxicity (liver, lung, eye, thyroid) |
DDI with CYP3A4 inhibitors (cyclosporine, some statins such as lovastatin or simvastatin), dose reduction during combining with warfarin or digoxin |
|
Dronedarone |
Bradyarrhythmia, QT prolongation, gastrointestinal adverse effect, fatigue, asthenia, hepatotoxicity |
Contraindication (heart failure with EF<40%, combining with antifungal and macrolide antibiotic agents), DDI with CYP3A4 inhibitors (verapamil/diltiazem), dose reduction of digoxin, edoxaban, or dabigatran during concomitant therapy, avoid combining of rivaroxaban when CrCl <80 mL/min |
|
Sotalol |
QT prolongation, TdP, bradyarrhythmia, adverse effect related with β-blocking effect (fatigue, weakness, dizziness) |
Dose reduction in patients with renal insufficiency (half-dose in patients with chronic renal insufficiency, 25% dose when GFR <30 mL/min/1.73m2) |
Table 4.Recommended testing for monitoring of toxicities in patients receiving amiodarone
|
Type of test |
Monitoring |
|
Thyroid function test (free T4 and TSH) |
Baseline and every 6 mo |
|
Liver function test |
Baseline and every 6 mo |
|
Chest X-ray |
Baseline and then yearly |
|
Pulmonary function test (including DLCO) |
Baseline and repeat if there is a clinical concern for pulmonary toxicity |
|
Ophthalmologic evaluation |
At baseline, if visual impairment |
|
Electrocardiogram |
Baseline and every year (at least) |
REFERENCES
- 1. Kirchhof P, Camm AJ, Goette A, Brandes A, Eckardt L, Elvan A, et al. Early rhythm-control therapy in patients with atrial fibrillation. N Engl J Med 2020;383:1305–16.ArticlePubMed
- 2. Tsadok MA, Jackevicius CA, Essebag V, Eisenberg MJ, Rahme E, Humphries KH, et al. Rhythm versus rate control therapy and subsequent stroke or transient ischemic attack in patients with atrial fibrillation. Circulation 2012;126:2680–7.ArticlePubMed
- 3. Coleman JJ, Pontefract SK. Adverse drug reactions. Clin Med (Lond) 2016;16:481–5.ArticlePubMedPMC
- 4. Vaughan Williams EM. A classification of antiarrhythmic actions reassessed after a decade of new drugs. J Clin Pharmacol 1984;24:129–47.ArticlePubMed
- 5. Lei M, Wu L, Terrar DA, Huang CL. Modernized classification of cardiac antiarrhythmic drugs. Circulation 2018;138:1879–96.ArticlePubMed
- 6. Task Force of the Working Group on Arrhythmias of the European Society of Cardiology. The Sicilian gambit: a new approach to the classification of antiarrhythmic drugs based on their actions on arrhythmogenic mechanisms. Circulation 1991;84:1831–51.ArticlePubMed
- 7. Dan GA, Martinez-Rubio A, Agewall S, Boriani G, Borggrefe M, Gaita F, et al. Antiarrhythmic drugs-clinical use and clinical decision making: a consensus document from the European Heart Rhythm Association (EHRA) and European Society of Cardiology (ESC) Working Group on Cardiovascular Pharmacology, endorsed by the Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS) and International Society of Cardiovascular Pharmacotherapy (ISCP). Europace 2018;20:731–732an.ArticlePubMed
- 8. Friedman PL, Stevenson WG. Proarrhythmia. Am J Cardiol 1998;82(8A):50N–58N.Article
- 9. Maisel WH, Kuntz KM, Reimold SC, Lee TH, Antman EM, Friedman PL, et al. Risk of initiating antiarrhythmic drug therapy for atrial fibrillation in patients admitted to a university hospital. Ann Intern Med 1997;127:281–4.ArticlePubMed
- 10. Hondeghem LM, Katzung BG. Antiarrhythmic agents: the modulated receptor mechanism of action of sodium and calcium channel-blocking drugs. Annu Rev Pharmacol Toxicol 1984;24:387–423.ArticlePubMed
- 11. Ikeda N, Singh BN, Davis LD, Hauswirth O. Effects of flecainide on the electrophysiologic properties of isolated canine and rabbit myocardial fibers. J Am Coll Cardiol 1985;5(2 Pt 1):303–10.Article
- 12. Kim M, Kwon CH, Lee JH, Hwang KW, Choi HO, Kim YG, et al. Right bundle branch block-type wide QRS complex tachycardia with a reversed R/S complex in lead V6: development and validation of electrocardiographic differentiation criteria. Heart Rhythm 2021;18:181–8.ArticlePubMed
- 13. Morganroth J. Risk factors for the development of proarrhythmic events. Am J Cardiol 1987;59:32E–37E.Article
- 14. Stanton MS, Prystowsky EN, Fineberg NS, Miles WM, Zipes DP, Heger JJ. Arrhythmogenic effects of antiarrhythmic drugs: a study of 506 patients treated for ventricular tachycardia or fibrillation. J Am Coll Cardiol 1989;14:209–17.ArticlePubMed
- 15. Falk RH. Flecainide-induced ventricular tachycardia and fibrillation in patients treated for atrial fibrillation. Ann Intern Med 1989;111:107–11.ArticlePubMed
- 16. Josephson ME. Antiarrhythmic agents and the danger of proarrhythmic events. Ann Intern Med 1989;111:101–3.ArticlePubMed
- 17. Passman R, Kadish A. Polymorphic ventricular tachycardia, long Q-T syndrome, and torsades de pointes. Med Clin North Am 2001;85:321–41.ArticlePubMed
- 18. Roden DM. Risks and benefits of antiarrhythmic therapy. N Engl J Med 1994;331:785–91.ArticlePubMed
- 19. Kay GN, Plumb VJ, Arciniegas JG, Henthorn RW, Waldo AL. Torsade de pointes: the long-short initiating sequence and other clinical features: observations in 32 patients. J Am Coll Cardiol 1983;2:806–17.ArticlePubMed
- 20. Jackman WM, Friday KJ, Anderson JL, Aliot EM, Clark M, Lazzara R. The long QT syndromes: a critical review, new clinical observations and a unifying hypothesis. Prog Cardiovasc Dis 1988;31:115–72.ArticlePubMed
- 21. Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehmann MH. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA 1993;270:2590–7.ArticlePubMed
- 22. Meierhenrich R, Helguera ME, Kidwell GA, Tebbe U. Influence of amiodarone on QT dispersion in patients with life-threatening ventricular arrhythmias and clinical outcome. Int J Cardiol 1997;60:289–94.ArticlePubMed
- 23. Grimm W, Steder U, Menz V, Hoffmann J, Maisch B. Effect of amiodarone on QT dispersion in the 12-lead standard electrocardiogram and its significance for subsequent arrhythmic events. Clin Cardiol 1997;20:107–10.ArticlePubMedPMC
- 24. Hondeghem LM, Snyders DJ. Class III antiarrhythmic agents have a lot of potential but a long way to go: reduced effectiveness and dangers of reverse use dependence. Circulation 1990;81:686–90.ArticlePubMed
- 25. Selzer A, Wray HW. Quinidine syncope: paroxysmal ventricular fibrillation occurring during treatment of chronic atrial arrhythmias. Circulation 1964;30:17–26.ArticlePubMed
- 26. Touboul P, Brugada J, Capucci A, Crijns HJ, Edvardsson N, Hohnloser SH. Dronedarone for prevention of atrial fibrillation: a dose-ranging study. Eur Heart J 2003;24:1481–7.ArticlePubMed
- 27. Hohnloser SH, Crijns HJ, van Eickels M, Gaudin C, Page RL, Torp-Pedersen C, et al. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009;360:668–78.ArticlePubMed
- 28. Le Heuzey JY, De Ferrari GM, Radzik D, Santini M, Zhu J, Davy JM. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J Cardiovasc Electrophysiol 2010;21:597–605.ArticlePubMed
- 29. Ruskin JN, McGovern B, Garan H, DiMarco JP, Kelly E. Antiarrhythmic drugs: a possible cause of out-of-hospital cardiac arrest. N Engl J Med 1983;309:1302–6.ArticlePubMed
- 30. Minardo JD, Heger JJ, Miles WM, Zipes DP, Prystowsky EN. Clinical characteristics of patients with ventricular fibrillation during antiarrhythmic drug therapy. N Engl J Med 1988;319:257–62.ArticlePubMed
- 31. Tisdale JE, Chung MK, Campbell KB, Hammadah M, Joglar JA, Leclerc J, et al. Drug-induced arrhythmias: a scientific statement from the American Heart Association. Circulation 2020;142:e214–33.ArticlePubMed
- 32. Barekatain A, Razavi M. Antiarrhythmic therapy in atrial fibrillation: indications, guidelines, and safety. Tex Heart Inst J 2012;39:532–4.PubMedPMC
- 33. Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo: the Cardiac Arrhythmia Suppression Trial. N Engl J Med 1991;324:781–8.ArticlePubMed
- 34. Pfisterer M. Negative inotropic effects of antiarrhythmic drugs: a clinical point of view. J Cardiovasc Pharmacol 1991;17 Suppl 6:S44–7.PubMed
- 35. El Hadidi S, Rosano G, Tamargo J, Agewall S, Drexel H, Kaski JC, et al. Potentially inappropriate prescriptions in heart failure with reduced ejection fraction: ESC position statement on heart failure with reduced ejection fraction-specific inappropriate prescribing. Eur Heart J Cardiovasc Pharmacother 2022;8:187–210.ArticlePubMedPDF
- 36. MacNeil DJ, Davies RO, Deitchman D. Clinical safety profile of sotalol in the treatment of arrhythmias. Am J Cardiol 1993;72:44A–50A.ArticlePubMed
- 37. Hamer AW, Arkles LB, Johns JA. Beneficial effects of low dose amiodarone in patients with congestive cardiac failure: a placebo-controlled trial. J Am Coll Cardiol 1989;14:1768–74.ArticlePubMed
- 38. Pfisterer M, Burkart F, Muller-Brand J, Kiowski W. Important differences between short- and long-term hemodynamic effects of amiodarone in patients with chronic ischemic heart disease at rest and during ischemia-induced left ventricular dysfunction. J Am Coll Cardiol 1985;5:1205–11.ArticlePubMed
- 39. Doshi D, Jayawardana R. Amiodarone-induced life-threatening refractory hypotension. Am J Case Rep 2015;16:617–20.ArticlePubMedPMC
- 40. Maghrabi K, Uzun O, Kirsh JA, Balaji S, Von Bergen NH, Sanatani S. Cardiovascular collapse with intravenous amiodarone in children: a multi-center retrospective cohort study. Pediatr Cardiol 2019;40:925–33.ArticlePubMedPDF
- 41. Naccarelli GV, Jalal S. Intravenous amiodarone: another option in the acute management of sustained ventricular tachyarrhythmias. Circulation 1995;92:3154–5.ArticlePubMed
- 42. Kober L, Torp-Pedersen C, McMurray JJ, Gotzsche O, Levy S, Crijns H, et al. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008;358:2678–87.ArticlePubMed
- 43. Connolly SJ, Camm AJ, Halperin JL, Joyner C, Alings M, Amerena J, et al. Dronedarone in high-risk permanent atrial fibrillation. N Engl J Med 2011;365:2268–76.ArticlePubMed
- 44. Gautier P, Guillemare E, Marion A, Bertrand JP, Tourneur Y, Nisato D. Electrophysiologic characterization of dronedarone in guinea pig ventricular cells. J Cardiovasc Pharmacol 2003;41:191–202.ArticlePubMed
- 45. Lim KS, Cho JY, Jang IJ, Kim BH, Kim J, Jeon JY, et al. Pharmacokinetic interaction of flecainide and paroxetine in relation to the CYP2D6*10 allele in healthy Korean subjects. Br J Clin Pharmacol 2008;66:660–6.ArticlePubMedPMC
- 46. Lewis GP, Holtzman JL. Interaction of flecainide with digoxin and propranolol. Am J Cardiol 1984;53:52B–57B.ArticlePubMed
- 47. Shea P, Lal R, Kim SS, Schechtman K, Ruffy R. Flecainide and amiodarone interaction. J Am Coll Cardiol 1986;7:1127–30.ArticlePubMed
- 48. Samlowski WE, Frame RN, Logue GL. Flecanide-induced immune neutropenia: documentation of a hapten-mediated mechanism of cell destruction. Arch Intern Med 1987;147:383–4.ArticlePubMed
- 49. Kuhlkamp V, Haasis R, Seipel L. Flecainide-induced hepatitis. Z Kardiol 1988;77:678–80.PubMed
- 50. Caron J, Libersa C. Adverse effects of class I antiarrhythmic drugs. Drug Saf 1997;17:8–36.ArticlePubMed
- 51. Kates RE, Yee YG, Kirsten EB. Interaction between warfarin and propafenone in healthy volunteer subjects. Clin Pharmacol Ther 1987;42:305–11.ArticlePubMed
- 52. Calvo MV, Martin-Suarez A, Martin Luengo C, Avila C, Cascon M, Dominguez-Gil Hurle A. Interaction between digoxin and propafenone. Ther Drug Monit 1989;11:10–5.ArticlePubMed
- 53. Gaita F, Richiardi E, Bocchiardo M, Asteggiano R, Pinnavaia A, Di Leo M, et al. Short- and long-term effects of propafenone in ventricular arrhythmias. Int J Cardiol 1986;13:163–70.ArticlePubMed
- 54. Guindo J, Rodriguez de la Serna A, Borja J, Oter R, Jane F, Bayes de Luna A. Propafenone and a syndrome of the lupus erythematosus type. Ann Intern Med 1986;104:589. ArticlePubMed
- 55. Mondardini A, Pasquino P, Bernardi P, Aluffi E, Tartaglino B, Mazzucco G, et al. Propafenone-induced liver injury: report of a case and review of the literature. Gastroenterology 1993;104:1524–6.ArticlePubMed
- 56. Pollak PT, Bouillon T, Shafer SL. Population pharmacokinetics of long-term oral amiodarone therapy. Clin Pharmacol Ther 2000;67:642–52.ArticlePubMed
- 57. McDonald MG, Au NT, Rettie AE. P450-based drug-drug interactions of amiodarone and its metabolites: diversity of inhibitory mechanisms. Drug Metab Dispos 2015;43:1661–9.ArticlePubMedPMC
- 58. Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001;40:587–603.ArticlePubMed
- 59. Sanoski CA, Bauman JL. Clinical observations with the amiodarone/warfarin interaction: dosing relationships with long-term therapy. Chest 2002;121:19–23.ArticlePubMed
- 60. Chitwood KK, Abdul-Haqq AJ, Heim-Duthoy KL. Cyclosporine-amiodarone interaction. Ann Pharmacother 1993;27:569–71.ArticlePubMedPDF
- 61. Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet 1997;350:1417–24.ArticlePubMed
- 62. Papiris SA, Triantafillidou C, Kolilekas L, Markoulaki D, Manali ED. Amiodarone: review of pulmonary effects and toxicity. Drug Saf 2010;33:539–58.ArticlePubMed
- 63. Lewis JH, Ranard RC, Caruso A, Jackson LK, Mullick F, Ishak KG, et al. Amiodarone hepatotoxicity: prevalence and clinicopathologic correlations among 104 patients. Hepatology 1989;9:679–85.ArticlePubMed
- 64. Boriani G, Blomstrom-Lundqvist C, Hohnloser SH, Bergfeldt L, Botto GL, Capucci A, et al. Safety and efficacy of dronedarone from clinical trials to real-world evidence: implications for its use in atrial fibrillation. Europace 2019;21:1764–75.ArticlePubMedPDF
- 65. Goldschlager N, Epstein AE, Naccarelli GV, Olshansky B, Singh B, Collard HR, et al. A practical guide for clinicians who treat patients with amiodarone: 2007. Heart Rhythm 2007;4:1250–9.ArticlePubMed
- 66. Mar PL, Horbal P, Chung MK, Dukes JW, Ezekowitz M, Lakkireddy D, et al. Drug interactions affecting antiarrhythmic drug use. Circ Arrhythm Electrophysiol 2022;15:e007955.ArticlePubMed
- 67. Naccarelli GV, Wolbrette DL, Levin V, Samii S, Banchs JE, Penny-Peterson E, et al. Safety and efficacy of dronedarone in the treatment of atrial fibrillation/flutter. Clin Med Insights Cardiol 2011;5:103–19.ArticlePubMedPMCPDF
- 68. Vallakati A, Chandra PA, Pednekar M, Frankel R, Shani J. Dronedarone-induced digoxin toxicity: new drug, new interactions. Am J Ther 2013;20:e717–9.ArticlePubMed
- 69. Pradaxa (dabigatran etexilate mesylate). Boehringer Ingelheim; 2018.
- 70. Savaysa (edoxaban) prescribing information. Daiichi Sankyo; 2015 [cited 2019 Nov 15]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206316lbl.pdf
- 71. Xarelto (rivaroxban). Janssen Pharmaceutical; 2011.
- 72. Wiggins BS, Dixon DL, Neyens RR, Page RL 2nd, Gluckman TJ. Select drug-drug interactions with direct oral anticoagulants: JACC review topic of the week. J Am Coll Cardiol 2020;75:1341–50.ArticlePubMed
- 73. Hoffmeister HM, Beyer M, Seipel L. Hemodynamic effects of the D- and L-isomers of sotalol on normal myocardium. Cardiovasc Drugs Ther 1991;5:1027–33.ArticlePubMedPDF
- 74. Antonaccio MJ, Gomoll A. Pharmacologic basis of the antiarrhythmic and hemodynamic effects of sotalol. Am J Cardiol 1993;72:27A–37A.ArticlePubMed
- 75. Orlando R, Piccoli P, De Martin S, Padrini R, Floreani M, Palatini P. Cytochrome P450 1A2 is a major determinant of lidocaine metabolism in vivo: effects of liver function. Clin Pharmacol Ther 2004;75:80–8.ArticlePubMed
- 76. Feely J, Wade D, McAllister CB, Wilkinson GR, Robertson D. Effect of hypotension on liver blood flow and lidocaine disposition. N Engl J Med 1982;307:866–9.ArticlePubMed
- 77. Rademaker AW, Kellen J, Tam YK, Wyse DG. Character of adverse effects of prophylactic lidocaine in the coronary care unit. Clin Pharmacol Ther 1986;40:71–80.ArticlePubMed
- 78. Caporaso NE, Shaw GL. Clinical implications of the competitive inhibition of the debrisoquin-metabolizing isozyme by quinidine. Arch Intern Med 1991;151:1985–92.ArticlePubMed
- 79. Kaukonen KM, Olkkola KT, Neuvonen PJ. Itraconazole increases plasma concentrations of quinidine. Clin Pharmacol Ther 1997;62:510–7.ArticlePubMed
- 80. Zhou HH, Anthony LB, Roden DM, Wood AJ. Quinidine reduces clearance of (+)-propranolol more than (–)-propranolol through marked reduction in 4-hydroxylation. Clin Pharmacol Ther 1990;47:686–93.ArticlePubMed
- 81. Vitali Serdoz L, Rittger H, Furlanello F, Bastian D. Quinidine: a legacy within the modern era of antiarrhythmic therapy. Pharmacol Res 2019;144:257–63.ArticlePubMed
Citations
Citations to this article as recorded by




 PubReader
PubReader ePub Link
ePub Link Cite
Cite

